MCPH1基因變異致原發性小頭畸形患兒的表型及遺傳學分析*
2024-03-25 09:22:38吳婉悅馬春元
罕少疾病雜志 2024年3期
吳婉悅 劉 毓 陳 凱 文 靜 馬春元 楊 瑩,*
1.貴陽市婦幼保健院罕見病中心內分泌遺傳代謝科 (貴州 貴陽 550001)
2.貴州省職工醫院消化內科 (貴州 貴陽 550025)
原發性小頭畸形(congenital microcephaly)又稱為真性小頭畸形或常染色體隱性遺傳小頭畸形,是以頭圍小、面部畸形和智力障礙為特征的神經系統發育性的罕見遺傳性疾病[1]。患兒的智商值取決于原發性小頭畸形的嚴重程度,一般為正常人平均值的30%~80%。不同地區的原發性小頭畸形發病率不同,為1/30 000~1/250 000,在近親結婚的人群中普遍較高[2]。目前已發現29個基因位點與原發性小頭畸形有關。MCPH1是第1個被鑒定的原發性小頭畸形致病基因[3-4]。本研究擬對1例小頭畸形、精神運動及認知發育落后患兒進行臨床分析和基因檢測,以明確病因。
1 材料和方法
1.1 研究對象患兒,男,2歲8個月,因發現頭小、運動及認知發育落后2年多就診。患兒3月齡時抬頭不穩,4月齡時家長發現患兒頭較同齡兒小伴運動及認知發育落后,表現為4月齡時豎頸不穩,喜歡打挺,不能逗笑,6月齡能翻身,8月齡雙手拇指內收握拳,視物主動抓物差,不能換手,不能獨坐,1歲時不能坐穩,不會發“爸爸媽媽”音,1歲10個月時會站立,2歲1個月會行走。現會喊“爸爸媽媽”,不會說短句,會拿勺吃飯,能獨立上下樓梯,不會單腳跳,不會跑。無抽搐、不自主抖動,無頭痛、視物模糊,無尖叫、雙目凝視等不適。曾就診于重慶醫科大學附屬兒童醫院,診斷為“全面性發育遲緩、原發性小頭畸形”,現予持續康復治療中。患兒系第3胎第2產,孕38+5周,剖宮產,出生體重3.1 kg,出生后無窒息及搶救史。母親為高齡產婦(36歲),孕早期有甲狀腺功能減退,予“左旋甲狀腺素片”口服,甲狀腺功能維持正常,現復查甲狀腺功能正常。第1胎因個人原因人工流產,第2胎5歲,姐姐,健康。父母體檢,非近親婚配,否認家族其他成員有類似病史。
1.2 方法
1.2.1 樣本采集 經患兒父母簽署知情同意書后,采集先證者及其直系親屬(父親,母親,姐姐)靜脈血各2 mL于乙二胺四乙酸(EDTA)抗凝管中(紫色),4℃ 冷藏。本研究經患兒監護人知情同意,醫院醫學倫理委員會批準(CN31-1915/R)。
1.2.2 DNA抽提 采用FlexiGene DNA Kit(Qiagen, Germany)提取受試者血液樣本基因組DNA。
1.2.3 突變檢測 DNA樣品送北京康旭醫學檢驗所檢測。采用IDT xGen Exome Research Panel V1.0 探針試劑盒(覆蓋人類基因組39 M)進行全外顯子捕獲,運用NovaSeq 6000平臺(Illumina,USA)進行測序,平均測序深度為100X,目標序列測序覆蓋度≥99%。獲得fastq 格式原始數據后,使用工具BWA(v0.7.15)將測序序列比對到人類參考基因組(Homo sapiens GRCh37)。參考1000G,dbSNP,ESP6500,ExAC等數據庫注釋患者變異在人群當中的頻率。使用GATK (v4) 分析 SNV(單核苷酸變異)和InDel(小的插入缺失變異)。使用 CODEX,XHMM(v1.0), EXOMEDEPTH,CLINCNV 和 KSCNV(康旭,北京)分析CNV(拷貝數變異),根據患兒的臨床表征尋找可疑致病性變異在HGMD Pro、PubMed和ClinVar數據庫中檢索變異的相關報道。采用Polyphen_2(Polymorphism Phenotyping v2)、SIFT (Sorting Intolerant From Tolerant)、Mutation Taster預測變異的致病性。根據美國醫學遺傳學和基因組學學會(American College of Medical Genetics and Genomics,ACMG)指南[8]對變異進行致病性評級。針對符合患兒表征的致病變異,使用ABI 3730測序系統(ABI,USA)在家系中驗證。
2 結 果
2.1 患兒一般臨床資料分析特殊面容:小頭畸形、下頜稍小、高腭弓、眼眥稍上斜,見圖1。體格檢查:體質量9.7 kg[低于同年齡、同性別、同地區幼兒平均體質量2個標準差(<-2 sd)],身高83.2 cm[低于同年齡、同性別、同地區幼兒平均身高2個標準差(<-2 sd)],頭圍46.0 cm[低于同年齡、同性別、同地區幼兒平均頭圍2個標準差(<-2 sd)],Tanner分期G1PH1,雙上肢肌力正常,雙下肢肌力Ⅳ級。雙下肢肌張力增高,緊張或哭吵時有角弓反張樣表現,左膝關節有彈響。膝反射:左側++、右側++。腹壁反射引出,巴氏征(-)、克氏征(-)、布氏征(-)。頭顱電子計算機斷層掃描(computed tomography,CT)示腦白質密度稍減低,雙側額葉腦溝、基底節區高密度。頭顱磁共振成像、頭顱磁共振彌散加權成像正常,腦電圖正常。Gesell發育診斷量表:個人社交相當于16.9周(發育商development quotient,DQ 48.4分),適應性相當于16周(DQ 45.8分),粗大運動相當于16周(DQ 45.8分),精細運動相當于12.0周(DQ 34.4分)語言能力相當于10.7周(DQ 30.7分)。兒童運動發育評估量表(粗大運動功能評估 Gross Motor Function Measure,GMFM):患兒粗大運動相當于4月齡水平,建議加強粗大運動功能訓練。兒童運動發育評估量表(精細運動能力評定表 Fine motorfunction measure, FMFM)):患兒精細運動功能發育水平較同齡兒落后,相當于3月齡水平,建議加強精細運動功能訓練。

圖1 患兒特殊面容、圖2 全外顯子測序與Sanger測序結。圖2A:先證者c.2312C>G雜合突變在父親基因組中未檢測到;圖2B:先證者c.445G>A雜合突變在父親基因組中驗證;圖2C:先證者c.2312C>G雜合突變在母親基因組中驗證,姐姐基因組未驗證;圖2D:先證者c.445G>A雜合突變在母親和姐姐基因組中均未驗證。
2.2 突變檢測及驗證結果全外顯子測序(WES)結果顯示,在先癥者MCPH1基因第6外顯子上發現錯義突變c.445G>A,該突變會使纈氨酸被異亮氨酸替換(Val149Lle);第13外顯子上發現另一錯義突變c.2312C>G,同樣會導致脯氨酸被精氨酸替換(Pro771Arg)。對先證者及其父母和姐姐進行Sanger 驗證,結果見表1及圖2。先證者父親攜帶MCPH1基因c.445G>A變異,未檢測到c.2312C>G。母親則攜帶MCPH1基因c.2312C>G變異,未檢測到c.445G>A;2個變異姐姐均未檢測到。至此,推測先證者MCPH1基因上發現的2個錯義突變(c.445G>A,c.2312C>G)可能分別源自父親和母親,遺傳模式如圖3所示。

表1 突變檢測
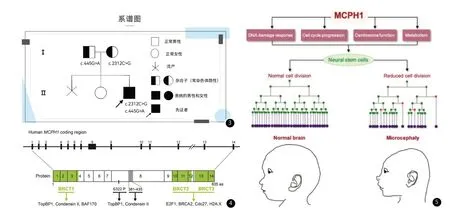
圖3 家系圖譜。圖4 MCPH1基因編碼序列結構示意圖。圖5 MCPH1 在大腦皮層發育中對神經祖細胞的調控示意圖。
2.3 變異位點致病性分析檢索HGMD Pro、PubMed和ClinVar數據庫,未發現MCPH1基因c.445G>A和c.2312C>G變異的報道(PM1)。ExAC數據庫和千人基因組數據庫中未見MCPH1基因c.445G>A變異和c.2312C>G變異被收錄(PM2)。根據OMIM數據庫描述,先證者表型高度符合MCPH1基因變異引起的MCPH(PP4)。采用Polyphen_2、SIFT、Mutation Taster預測變異致病性在線軟件預測MCPH1基因c.445G>A變異為良性(BH4),MCPH1基因c.2312C>G變異為有害(PP3)。根據ACMG指南,綜合判斷MCPH1基因c.445G>A為可能致病變異(PM1+PM2+PP4+BP4),MCPH1基因c.2312C>G亦為可能致病變(PM1+PM2+PP4+PP3)。
3 討 論
小頭畸形分為原發性小頭畸形和繼發性小頭畸形。原發性小頭畸形疾病是一種罕見的神經分裂異常引起的腦發育疾病,病人主要臨床特征為頭圍減小并伴隨不同程度的智力衰退,疾病分型較多,OMIM數據庫收錄原發性小頭畸形共29個分型,其中原發性小頭畸形(1 型 )屬于常染色體隱性遺傳(AR,OMIM: 251200),占比約19%。該疾病的致病基因是MCPH1(8p23.1)[3-4]。MCPH1基因編碼的MCPH1蛋白是一種神經發育系統疾病的相關蛋白,與細胞分裂有關,常見于神經系統中。當MCPH1基因突變,編碼蛋白受阻,干擾中心體或紡錘體的正常形成,影響細胞周期及 DNA損傷修復等過程,進而影響神經前體細胞的增殖、分化和凋亡等過程,最終導致神經元數量減少并形成偏小的大腦[5-8]。
MCPH1基因含14個外顯子(圖4),其編碼蛋白含有3個BRCT結構域,N端含有1個BRCT結構域,C端含有2個串聯BRCT結構域,分別為BRCT2和BRCT3,第8外顯子編碼的376-485個氨基酸形成的結構域可以與凝縮蛋白II相互作用[7,9]。MCPH1通過結合不同的因子在細胞周期和DNA損傷修復中發揮作用。MCPH1基因調節大腦大小,在胚胎大腦發育過程中用于調節神經干細胞分裂模式相關的許多細胞功能。由于神經干細胞的過早分化(圖五,紅點表示)和自我更新分裂的減少(圖五,綠點表示)以及功能受損導致神經元生成不足(圖5,深紫色點表示),受影響的個體會出現小頭畸形[8,10](圖5)。本例報告患兒基因致病證據不充分,生物信息學蛋白質功能預測MCPH1基因c.445G>A變異為良性,MCPH1基因c.2312C>G變異為有害。但與患兒臨床表型高度符合,故考慮除MCPH1基因(8p23.1)區域突變可導致原發性小頭畸形疾病,是否存在影響功能內含子區域的突變,還需進一步驗證。
