納米氧化鋅液相法制備技術進展
2024-03-25 09:10:06李春雷田玉琴
無機鹽工業 2024年3期
楊 卓,李春雷,張 鑫,喬 勉,田玉琴,宮 源
(1.蘭州理工大學石油化工學院,甘肅蘭州 730050;2.蘭州理工大學甘肅省低碳能源化工重點實驗室,甘肅蘭州 730050)
納米氧化鋅至少有一維尺寸介于1~100 nm,由于其晶粒的納米化,表面電子分布和晶體結構發生改變,產生了微米尺度氧化鋅所不具備的良好的表面效應、體積效應、熱效應及化學穩定性[1-3]。基于上述優點,納米氧化鋅廣泛應用于橡膠[4-5]、涂料[6-7]、催化[8-10]、半導體[11-13]、陶瓷[14]和紡織[15-16]等領域。其中,在橡膠領域的應用最為重要,可替代普通活性氧化鋅作為橡膠輪胎硫化工藝中重要的活化劑,在保證硫化后橡膠耐磨性、抗撕裂性、拉伸強度等優異物理性能的同時,還可以顯著降低用量至普通活性氧化鋅用量的30%~50%,大幅降低橡膠輪胎使用中產生的鋅污染[4]。數據顯示,2022 年中國橡膠外胎累計產量達8.56 億條,按每條外胎鋅用量為2.5%(質量分數)計算,2022年國內納米氧化鋅需求量約為62萬t,市場前景廣闊。
近幾十年來,納米氧化鋅制備技術發展迅速,按反應體系相態分類,主要包括氣相法、固相法和液相法[17-18]。其中液相法因工藝條件易控制、所得產品純度更高、技術成本更低,相比于其他兩類方法更適用于工業規模化生產。液相法的核心思路是將可溶性鋅鹽與沉淀劑在一定條件下混合,形成液相反應體系。體系中的鋅離子與沉淀劑陰離子相互作用,直接或間接生成納米氧化鋅。根據反應體系不同,液相法可細分為微乳液法、溶膠-凝膠法、水熱/溶劑熱法和化學沉淀法。不同方法所得納米氧化鋅形貌、平均粒徑及粒徑分布受體系及具體工藝條件的影響而不同[19-20]。本文重點對上述納米氧化鋅液相制備方法的基本原理及關鍵影響因素進行了綜述,在此基礎上介紹了一種利用氣泡間液膜厚度調控顆粒尺寸,以制備納米氧化鋅的連續液相法——“氣泡液膜法”。
1 微乳液法
微乳液是指兩種互不相溶的液體在表面活性劑的作用下混合,形成存在液-液相界面(油相與水相)的液體分散體系,所分散的液滴尺寸為1~100 nm。利用微乳液法制備納米氧化鋅時,可溶性鋅鹽與沉淀劑分別分散在油相中形成微乳液后兩者混合成油包水型(W/O)微乳液反應體系。晶體在由表面活性劑和油相包圍的“水池”型微反應器中成核與生長。由于制備微乳液時需加入表面活性劑,微乳液法在一定程度上可以防止納米顆粒間的團聚,改善其分散性。影響微乳液法所得納米氧化鋅形貌及粒徑的關鍵因素包括以下3個方面。
1.1 物料配比的影響
SUN 等[21]通過改變醋酸鋅與氫氧化鈉物質的量比(w),并加入陰離子表面活性劑十二烷基硫酸鈉(SDS)形成微乳液反應體系經水熱處理形成了六方纖鋅礦型氧化鋅納米線,合成機理如圖1所示。研究了在不同w條件下(5、10、15、20、25)對產物氧化鋅形貌的影響,發現氧化鋅納米線的長徑比隨w 增大而增加,當w大于20時長徑比反而下降(見圖2)。
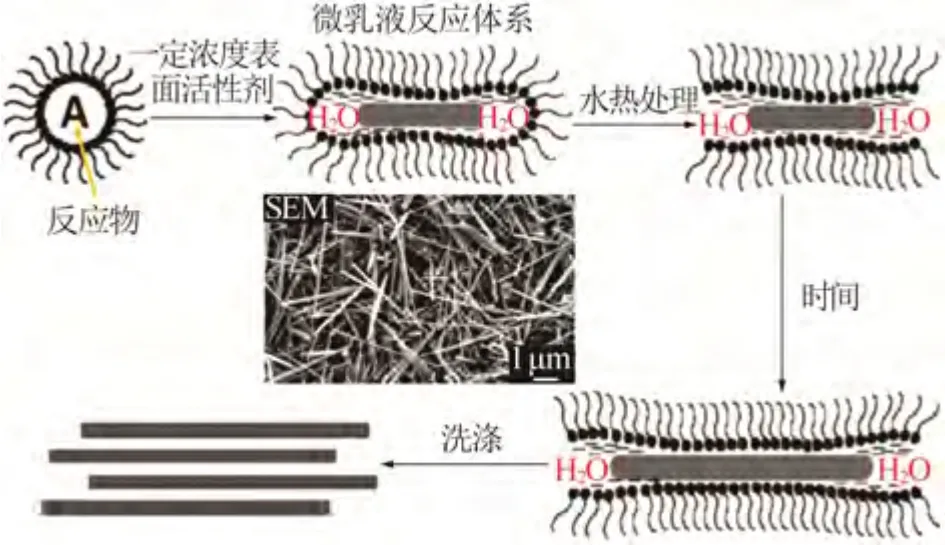
圖1 氧化鋅納米線形成機理圖[21]Fig.1 Schematic illustration of formation mechanism of ZnO nanowires[21]
DING 等[22]以六水合硝酸鋅溶液為水相、環己烷為連續油相、聚乙二醇辛基苯基醚(Triton X-100)為表面活性劑、正己醇為共表面活性劑制備了W/O型微乳液,結合煅燒處理制備了穿孔的氧化鋅納米片長為300~500 nm。氧化鋅納米片的尺寸可以通過調整水與非離子表面活性劑Triton X-100的物質的量比w(5、10、15、20、30、50)來控制。隨著w的變化,氧化鋅納米片的形態也從離散的顆粒到相互連接的納米顆粒,最后到致密的氧化鋅納米片。
1.2 表面活性劑種類的影響
LIM 等[23]探究了兩種表面活性劑乙苯酸鈉鹽(EBS)和十二烷基苯磺酸鈉鹽(DBS)對氧化鋅納米棒形貌的影響,兩者與醋酸鋅在二甲苯中形成微乳液反應體系于140 ℃下反應5 h;結果顯示,隨著表面活性劑烷基鏈長度的增加,納米棒變長加粗,長鏈的DBS 形成較大“水池”型微反應器導致氧化鋅納米棒平均直徑更大(300 nm)而長徑比較小。相反,EBS 所得納米棒長徑比更大且比表面積更高(平均直徑為80 nm),這可能是由于其微乳液體系的特性所致。
VAN DER RUL 等[24]構建了氯化鋅-二(2-乙基)己基磺化琥珀酸鈉(表面活性劑)-庚烷-氫氧化鈉四元微乳液反應體系,于70 ℃下反應得到平均粒徑約為12 nm,粒徑分布均一的納米氧化鋅。該研究特別指出,表面活性劑在產物表面的附著可有效維持納米顆粒間的分散,避免團聚現象。
1.3 添加劑種類的影響
LI等[25]使用硝酸鋅和氫氧化鈉,在微乳液中形成反應體系,經過140 ℃水熱處理15 h 制備了平均粒徑小于100 nm 的氧化鋅。選用聚乙二醇(PEG)為添加劑,研究了不同PEG濃度對氧化鋅形貌和尺寸的影響。結果顯示,未添加PEG 時,氧化鋅呈針狀或球狀,且存在顆粒團聚現象;隨著PEG 質量分數由12.5%升高至25.0%,氧化鋅由針狀轉變為棒狀,粒徑減小且分散更均勻;當PEG 質量分數達到50%時,氧化鋅顆粒變為球形,但再次出現顆粒團聚,上述變化見圖3。研究認為,添加劑在微乳液中能有效吸附在產物晶面上,從而控制納米晶體的生長方向,最終改變其形貌。

a—微乳液中的反應過程;b—0.0%;c—12.5%;d—25.0%;e—50.0%。圖3 PEG質量分數對氧化鋅形貌和粒徑的影響[25]Fig.3 Effect of PEG mass fraction on morphology and particle size of zinc oxide[25]
MOHAMAD 等[26]以六水合硝酸鋅為鋅源,添加劑為十六烷基三甲基溴化銨(CTAB)、100 mL正己烷作為油相,分別用溴35(0.004 mol)和1-丁醇(10 mL)作為表面活性劑和共表面活性劑制備微乳液。沉淀物經干燥、煅燒后制備出棒狀摻釩氧化鋅,粒子的尺寸在60~130 nm。其中通過改變添加劑CTAB 的加入量調控了產物形貌為立方體形或者棒狀,也促進了納米氧化鋅中釩的摻雜。
2 溶膠-凝膠法
溶膠-凝膠法制備納米顆粒的一般流程是利用含高化學活性組分的化合物,如金屬醇鹽或無機鹽作為前驅物,溶于液相后經水解、縮合反應,形成穩定的透明溶膠,溶膠經陳化后膠粒間緩慢聚合,形成三維空間網格結構的凝膠;最終凝膠經干燥、焙燒去除有機成分,得到無機納米材料。通過該方法制備納米氧化鋅最早由SPANHEL 等[27]報道,其采用的前驅物包括二水醋酸鋅、無水乙醇和氫氧化鋰。經過不斷發展,目前最常用的鋅源仍為二水醋酸鋅,溶膠及凝膠制備過程所用添加劑則更為多樣化。溶膠-凝膠法的優點是能更快地成核和生長,反應過程條件易控制,所得納米氧化鋅分散性好、純度高,但成本昂貴、造價較高[28]。
2.1 添加劑對溶膠制備和形貌的影響
GULER 等[29]將二水醋酸鋅于60 ℃下溶于乙二醇單甲醚中制備溶膠,保持溫度恒定,向所得溶膠中加入添加劑乙醇胺后繼續攪拌得到凝膠,再經600 ℃焙燒8 h 得到形貌呈球形、大小均一、平均粒徑小于100 nm 的納米氧化鋅。不同于上述報道,VAFAEE等[30]在溶膠制備過程中采用了三乙醇胺與乙醇的混合液作為添加劑,將二水醋酸鋅加入到上述混合液中,并于50~60 ℃下攪拌混合30 min,所得溶膠陳化1 ~ 2個月以探究其穩定性,并分析了二水醋酸鋅的加入量和三乙醇胺與醋酸鋅物質的量比對所得納米氧化鋅形貌及粒徑的影響。結果表明,當二水醋酸鋅濃度為0.25~0.75 mol/L,三乙醇胺與醋酸鋅物質的量比為3∶5 時,所得納米氧化鋅形貌呈球形,粒徑小至2~4 nm。通過傅里葉紅外光譜分析認為,上述溶膠形成過程中會發生如下化學反應:
二水醋酸鋅首先水解為帶正電荷的中間物單醋酸鋅[CH3COOZn]+,再與三乙醇胺縮合,形成新的含鋅化合物[CH3COOZn]N(CH2CH2OH)2。中間物單醋酸鋅是溶膠-凝膠法形成氧化鋅的關鍵。
JIANG 等[31]使用乙二胺替代三乙醇胺,將二水醋酸鋅、乙醇和乙二胺混合形成溶膠,然后加入草酸,經過濾、洗滌、煅燒制備出棒狀或針狀的納米氧化鋅。當乙二胺與二水醋酸鋅物質的量比為0.1時,產物呈現針狀,直徑為280~320 nm,長度為2.5~7 μm;當比例為0.2 時,產物尺寸減小呈棒狀,直徑為20~200 nm,長度為0.2~1.5 μm;比例增至0.5 時,產物更均勻直徑約為480 nm,長度為4.2 μm。這表明,乙二胺作為添加劑在溶膠制備中對納米氧化鋅形貌有關鍵影響。與三乙醇胺不同,乙二胺中的雙氮原子與鋅離子形成鰲合物,在制備過程中,這種鰲合物的分解控制了納米晶體的成核和生長過程,導致棒狀納米顆粒的形成。
2.2 凝膠后處理操作對粒徑的影響
SEID 等[32]探究了退火溫度對納米氧化鋅結晶度及晶粒大小的影響。其采用溶膠-凝膠法合成摻雜銦的棱柱狀、紡錘狀或圓盤狀納米氧化鋅,原料包括二水醋酸鋅、氫氧化鈉、硝酸銦和一乙醇胺[33]。產物經過過濾、水洗、干燥后進行退火處理;XRD分析顯示隨著退火溫度從300 ℃升高到800 ℃,納米氧化鋅結晶度提高,且基于Scherrer 公式計算的晶粒尺寸由25.5 nm增大至31.6 nm。
SIRIN 等[34]以二水醋酸鋅為主要原料,分別采用單乙醇胺(MEA)和2-甲氧基乙醇作為穩定劑和溶劑,二水醋酸鋅與MEA物質的量比保持在1∶1,最終制備出氧化鋅納米薄膜。在進行退火處理時,溫度從350 ℃提高到550 ℃,溫度梯度為50 ℃。通過XRD 譜圖分析發現,隨著退火溫度的升高,氧化鋅薄膜的結晶度有所提高。在溫度為400、450、500、550 ℃時,氧化鋅薄膜的平均晶體尺寸分別為11.18、12.67、22.20、16.49 nm。這可能是由于退火溫度相對較高時(>500 ℃),大部分間隙O原子進入晶格位取代Zn 原子,會導致嚴重的晶格畸變,在薄膜中引起較大的應力,影響氧化鋅晶體的正常生長。
3 水熱/溶劑熱法
水熱/溶劑熱法制備納米顆粒的特征在于反應組分在高溫、高壓液相封閉體系中進行化學反應,相比于常溫常壓條件,水熱反應的反應速率將顯著提高。水熱法以水作為介質,溶劑熱法則是在水熱法的基礎上發展起來,以有機物或者非水溶液為介質,擴大了水熱法的應用范圍。水熱法具有操作簡單、反應條件可控的優點,其合成納米顆粒的控制因素主要有水熱反應前驅物的種類、反應溫度、時間、體系pH、反應物濃度、反應介質、添加劑等。
3.1 水熱法
SAJID等[35]將二水醋酸鋅和十六烷基三甲基溴化銨(CTAB)溶于去離子水中充分混合,采用氫氧化鈉和鹽酸共同調節體系pH至8左右,之后轉入水熱釜中于180 ℃下反應24 h,得到寬度為150~300 nm,厚度為10~60 nm的板狀納米氧化鋅,且表面具有納米孔結構,孔徑約為10 nm,比表面積達到126 cm2/g。研究認為,CTAB 作為螯合劑參與水熱反應,對其氧化鋅形貌控制起關鍵作用。
FANG等[36]采用水熱法制備納米氧化鋅用作光催化劑,一組實驗向氫氧化鈉水溶液中滴加二水醋酸鋅溶液至沉淀消失,隨后加入CTAB,在160 ℃水熱反應12 h后得到花狀納米氧化鋅;另一組實驗以葡萄糖作為添加劑,加入二水醋酸鋅溶液,180 ℃水熱反應12 h 后制備出球形納米氧化鋅。由SEM 和TEM 照片分析顯示,花狀氧化鋅由圓錐尖的一維六方納米棱柱組成,直徑為100~400 nm;球形納米氧化鋅平均粒徑為2~3 μm,由直徑為100~250 nm的晶粒團聚形成(圖4);綜上所述,添加劑可以顯著影響水熱法制備的納米氧化鋅的形貌和粒徑大小。

a、b—花狀結構;c、d—球狀結構。圖4 納米氧化鋅的SEM和TEM照片[36]Fig.4 SEM and TEM images of nano zinc oxide[36]
3.2 溶劑熱法
ANZLOVAR 等[37]利用B-ZnCO3-1(平均粒徑為200 nm,粒狀)和B-ZnCO3-2(平均粒徑>1 μm,片狀)兩種堿式碳酸鋅為前驅物,以對甲苯磺酸(p-TSA)和二元醇為溶劑,通過溶劑熱法制備了納米氧化鋅。研究發現,溶劑中的p-TSA 還可以作為催化劑來提高堿式碳酸鋅在水相中的溶解度,加速水熱反應;B-ZnCO3-1 因具有較小的平均粒徑而表現出更高的水熱反應活性,當p-TSA 濃度為0.02 mol/L時,氧化鋅平均粒徑為150~250 nm分布較寬;p-TSA濃度提高至0.04 mol/L 時,粒徑減小為70~90 nm 且分布變窄;進一步增加p-TSA 濃度至0.08 mol/L,納米氧化鋅平均粒徑最終為50~60 nm。
TRYFON 等[38]將水合乙酰丙酮鋅溶解在4 mL的油胺(OAm)中,放入200 ℃的高壓釜中進行溶劑熱過程,制備的不規則顆粒狀納米氧化鋅結晶尺寸為19 nm,還發現OAm 作為溶劑使產物粒徑分布窄且顆粒尺寸較小,增強了納米氧化鋅的分散性和穩定性。
杜慶波等[39]以二水醋酸鋅為鋅源制取微納米級別的 ZnO,采用無水乙醇和十六烷基三甲基溴化銨(CTAB)為溶劑,通過改變反應時間和反應溫度,制備出簇狀、棒狀和球狀的氧化鋅;將此不同形貌的納米ZnO 作為催化劑處理含亞甲基藍溶液(MB)的模擬污水,驗證了其優異的光催化性能,降解率可達95.45%。
3.3 低溫水熱法
SUN 等[40]報道了一種低溫水熱合成超細氧化鋅納米線/納米管陣列的方法,該方法包括混合一定濃度的硝酸鋅和六亞甲基四胺 (HMT) 溶液,并將預制的氧化鋅薄膜包覆硅片作為基底置于混合液中,整個體系在90 ℃下密封并進行水熱恒溫反應數小時促使納米顆粒在基底上生長,經過9 h水熱反應并進行臨界點干燥處理后,所得氧化鋅呈現納米線陣列且排列整齊,單個顆粒軸向長度>2 μm,直徑≤20 nm,如圖5所示。

圖5 反應9 h后經臨界點干燥處理的氧化鋅SEM(a)和TEM(b)照片[40]Fig.5 SEM(a) and TEM(b) images of zinc oxide grown for 9 h and post-treated by critical point drying[40]
KUSUMAM等[41]將氫氧化鈉和十六烷基三甲基溴化銨(CTAB)混合,滴加0.1 mol/L 六水合硝酸鋅,將得到的溶液轉移到500 mL不銹鋼高壓釜中,并在90 ℃的真空烘箱中保存3 h 獲得白色沉淀物,再將其洗滌、干燥、煅燒制備出厚度小于30 nm的氧化鋅納米片。
4 化學沉淀法
化學沉淀法工藝最為簡單,將沉淀劑與可溶性鋅鹽混合,液相游離鋅離子與沉淀劑陰離子結合,直接或間接地形成納米氧化鋅;直接沉淀時,沉淀劑選擇強堿溶液如氫氧化鈉、氫氧化鉀、氫氧化鋰等在特定溫度下進行,而選擇弱堿溶液時,沉淀為氫氧化鋅或堿式碳酸鋅,需經過進一步熱分解間接制備納米氧化鋅;相比于前述的液相制備技術,化學沉淀法原料成本最低、使用設備簡單,可廣泛應用于大規模工業生產[42]。
4.1 影響化學沉淀過程的因素
HERRERA 等[43]采用田口實驗探究了鋅源、沉淀劑及溶劑種類、攪拌速率及時間、結晶過飽和度、Zn2+濃度、反應溫度等(見表1)對化學沉淀法所得氧化鋅形貌及粒徑大小的影響。所得氧化鋅呈六方纖鋅礦結構且形貌多樣,包括球狀、線狀、棒狀、花狀及四面體狀;田口實驗分析表明,除了反應溫度、攪拌速率和時間外,其他因素對納米氧化鋅粒徑影響也很顯著,尤其是結晶過飽和度;其中在90 ℃下醋酸鋅和碳酸銨反應,乙醇為溶劑,結晶過飽和度為25%,制備的線狀納米氧化鋅具有最小粒徑(34.7 nm)。
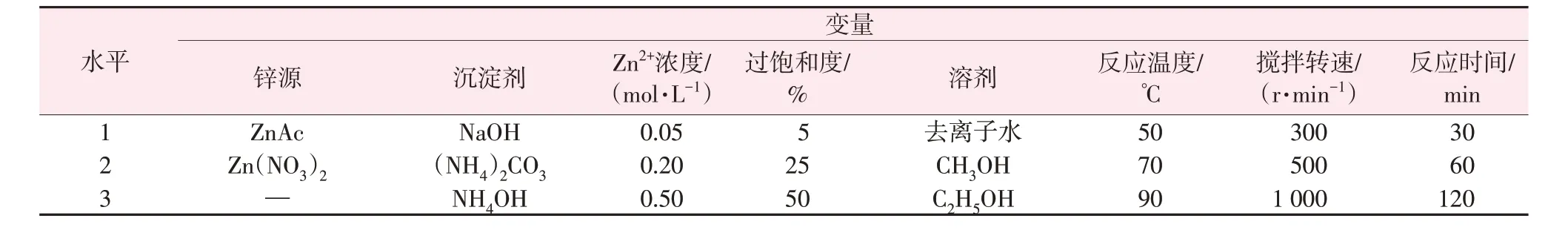
表1 合成氧化鋅的變量及其水平[43]Table 1 List of variables and levels used in synthesis of ZnO[43]
此外,直接沉淀時體系pH 對沉淀形式影響顯著,高pH有利于氫氧化鋅的形成,低pH則有利于氧化鋅的形成[44]。LU 等[45]將硝酸鋅溶液與沉淀劑六亞甲基四胺(HMT)溶液混合,反應過程如式(3)~(6)所示:
由于受pH和Zn2+濃度影響,反應(5)和(6)之間存在競爭。研究發現,在pH為6.8~7.0時,HMT與硝酸鋅物質的量比對ZnO 的形成、形貌和粒徑控制至關重要。當n(HMT)∶n(硝酸鋅)=1∶1 時,前驅體Zn(OH)2為小于100 nm 的無定形薄片狀(圖6);大于5∶1 時,高濃度HMT 將顯著阻礙OH-向新生沉淀表面的沉積,有利于ZnO 生成,且為了降低表面能,HMT 更傾向于吸附在高表面能的[0110]晶相,阻止了ZnO在該方向的生長,最終形成厚六邊形結構。于300 ℃下對n(HMT)∶n(硝酸鋅)=1∶1 制備的Zn(OH)2進行高溫陳化,得到平均粒徑為3.3 nm 的ZnO納米顆粒。
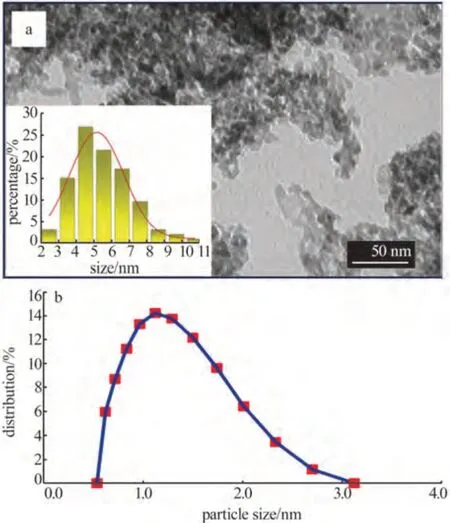
圖6 HMT與Zn(NO3)2物質的量比為1∶1時ZnO的TEM圖及粒徑分布圖(a)和DLS法測定了HMT與Zn(NO3)2物質的量比為1∶1時ZnO的粒徑(b)[45]Fig.6 TEM image of ZnO at n(HMT)∶n[Zn(NO3)2]=1∶1 and particle size distribution(a) and ZnO particle size at n(HMT)∶n[Zn(NO3)2]=1∶1 measured by DLS(b)[45]
SHARMA 等[46]將二水醋酸鋅和25%的氨水混合后在85 ℃下連續攪拌2 h,形成乳白色沉淀,體系內主要發生如式(7)~(9)反應:
經分析可知所得沉淀為氫氧化鋅和氧化鋅的混合物,對其進行水洗、干燥后再經300、400 ℃煅燒2 h得到球形或輕微扭曲的三角形納米氧化鋅,平均粒徑約為30 nm。此外,對比發現400 ℃煅燒后的產物粒徑分布更為均一。
POURRAHIMI 等[47]在60 ℃下,使用不同類型和濃度的鋅鹽溶液與NaOH 溶液快速混合,攪拌60 min直接制備納米氧化鋅,并研究了4種鋅鹽(二水醋酸鋅、氯化鋅、硫酸鋅和硝酸鋅)對產物粒徑和形貌的影響;結果顯示,二水醋酸鋅對納米粒子的穩定性最高,產物呈棱錐狀,粒徑分布最窄,平均粒徑為25 nm(圖7a);氯化鋅和硫酸鋅制備的納米氧化鋅粒徑分別為10~30 nm(圖7b)和80~100 nm、呈現花瓣狀結構(圖7c);使用硝酸鋅時,納米氧化鋅形貌呈現更大的星狀結構,由花瓣狀粒子聚集形成,平均粒徑約為500 nm(圖7d)。

a—二水醋酸鋅;b—氯化鋅;c—硫酸鋅;d—硝酸鋅;e—4種物質的體積密度分布。圖7 不同鋅鹽為原料所得氧化鋅形貌及顆粒數與體積的密度分布[47]Fig.7 Morphology and number/volume frequency distribution of zinc oxide formed from different zinc salts[47]
邸琬茗等[48]對化學沉淀法工藝進行了改進,以尿素為沉淀劑與硝酸鋅反應并維持體系過飽和度穩定,生成高結晶度、尺寸均一的堿式碳酸鋅前驅體,晶粒尺寸為25~30 nm。經450 ℃焙燒獲得最小粒徑為47.3 nm 的球形多孔型納米氧化鋅;研究發現,尿素分解是沉淀過程的控速步驟,提高溫度或尿素濃度可加速分解,增加前驅體的收率。分析發現,反應初期生成氧化鋅,但隨反應體系pH 的降低,會形成Zn4CO3(OH)6·H2O和Zn5(CO3)2(OH)6兩種形式的堿式碳酸鋅,并存在前者向后者轉化的現象。
4.2 過程強化對化學沉淀過程的影響
實現液相法納米氧化鋅制備技術的設備也是研究的熱點之一,WANG 等[49]采用流動注射合成技術(FIS),將氨基甲酸銨和氯化鋅兩種反應物同時注入設備反應管內的載流中混合,反應制備氧化鋅前驅體堿式碳酸鋅,反應方程如下:
流動注射合成器的優點是將沉淀反應限制在很短的時間間隔內(1~2 min),故可以顯著地限制初生顆粒間的團聚,且控制結晶過程以初級成核為主。通過該技術制備得到了納米尺度堿式碳酸鋅前驅體,在350 ℃煅燒3 h 后所得氧化鋅形貌呈棒狀,粒度為10~15 nm(見圖8)。

圖8 FIS系統示意圖和所得ZnO前驅體、煅燒后產物的SEM照片[49]Fig.8 Schematic representation of FIS system and SEM images of ZnO precursor and product after calcination[49]
從結晶動力學角度分析,高的結晶過飽和度可以促進成核的發生,有利于形成納米顆粒[50]。對于反應結晶,離子間的化學反應速率遠快于反應器內的傳質速率,強化傳質過程以達到均勻且高的結晶過飽和度對于納米顆粒的形成至關重要。而在傳統攪拌釜式反應器中,由于混合的不理想,特別是難以快速且均勻地實現分子尺度的微觀混合,得到尺度均一的納米顆粒十分困難。采用過程強化手段,提高反應體系過飽和度是制備納米顆粒的有效途徑。
微流控技術是重要的過程強化手段,在特征尺度幾十至幾百微米的微反應器內進行反應,可大幅提升物料的混合效率,實現對反應條件的精確控制[51]。清華大學駱廣生團隊應用自制的膜分散微反應器(如圖9a),在孔徑為5 μm的不銹鋼微孔濾膜分散下,以硫酸鋅和碳酸氫銨中的一種作為分散相,另一種作為連續相在微通道內快速且充分混合,實現高的結晶過飽和度;所得堿式碳酸鋅前驅體經300 ℃煅燒,得到窄分布且平均粒徑為9.33 nm,形貌呈類球狀的氧化鋅納米顆粒(見圖9b)[52]。超重力技術同樣可以顯著強化微觀混合,超重力反應器通過填料床層的高速旋轉模擬超重力環境,產生的巨大剪切力可以不斷破碎液流,產生超薄液膜、超細液絲及超小液滴,使得相際之間的接觸、微觀混合更加充分[53]。北京化工大學陳建峰院士團隊的黃謝君將超重力反應器成功用于納米氧化鋅的制備[54],采用醋酸鋅和氫氧化鉀的甲醇溶液,以超重力一步法合成氧化鋅,并通過添加四乙氧基硅烷對所得產物進行表面改性,實驗流程如圖9c 所示,最終得到平均粒徑為4 nm 的超細氧化鋅納米粒子(圖9d)。相比于釜式法,超重力法制備的納米氧化鋅同為六方晶型,但顆粒粒徑更小、分布更均勻,在液相介質中分散性更好,分散體的可見光透過率更高,由此體現了超重力法在制備更高品質納米材料上的優勢。
蘭州大學楊第倫等在化學沉淀法的基礎上提出了連續制備納米粒子的新方法“氣泡液膜法”,設計了以泡罩碟式攪拌盤為核心部件的氣泡液膜反應器[55-58]。泡罩碟式攪拌盤高速旋轉充氣,混合鋅鹽溶液、沉淀劑與表面活性劑,形成高密度微氣泡泡沫液,氣體為分散相、液體為連續相,液膜厚度最薄為10~100 nm(圖10)。氣泡液膜法調控液膜厚度可控制納米氧化鋅顆粒大小,并基于表面活性劑的自組裝得到疏松型產物,煅燒后的納米氧化鋅平均粒徑為30~50 nm,團聚指數為13,優于GB/T 19589—2004《納米氧化鋅》。
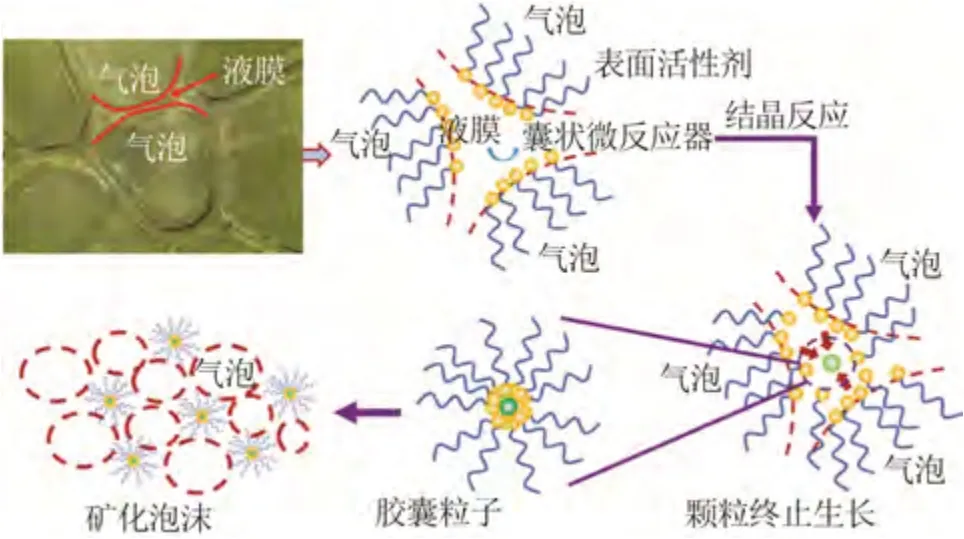
圖10 氣泡液膜法制備堿式碳酸鋅前驅體原理示意圖[55-58]Fig.10 Principle diagram of preparing precursor of basic zinc carbonate using bubble liquid film method[55-58]
除制備納米氧化鋅外,該方法已成功實現小試規模的納米碳酸鈣、超微紅磷復合阻燃劑、超微納米磷酸鐵等高附加值產品的制備。目前,蘭州蘭石中科納米科技有限公司與白銀有色集團股份有限公司達成合作意向,以白銀集團鋅冶煉工藝副產的含氯浮渣固廢為鋅源,利用氣泡液膜反應技術,建設5 000 t/a 納米氧化鋅生產示范線,產線建成后鋅浮渣固廢處理能力有望實現4 000 t/a。
5 結語與展望
本文重點對微乳液法、溶膠-凝膠法、水熱/溶劑熱法和化學沉淀法等多種納米氧化鋅液相法制備技術的基本原理及關鍵影響因素進行了綜述。化學沉淀法因原料成本低、設備相對簡單,在大規模工業生產上具有顯著優勢,各種過程強化技術,如微流控、超重力的應用,有效強化了反應物料間的微觀混合,極大限度地提高了結晶過飽和度,促進晶體成核而限制生長,實現粒子納米化。過程強化技術及設備的開發使化學沉淀法的工業生產規模更加擴大,所得產品品質更加穩定。因此,過程強化技術及其關鍵設備在納米氧化鋅制備中的應用,是未來行業發展的主要方向。
氣泡液膜法是建立在化學沉淀法基礎上的連續化納米氧化鋅制備的新方法,其通過控制液膜厚度,使成核晶體在液膜內限域生長,所得產品更為均一,且在一定程度上克服了納米粒子間易團聚的關鍵技術難題,具有較廣闊的工業應用前景。在現階段,深入掌握氣泡液膜反應環境中納米粒子的生長原理及動力學規律,對于反應器的進一步優化與放大具有重要指導意義。
