基于重測序數據評估南陽牛保種效果
2024-10-14 00:00:00劉思宇張曼張巖魏稚彤祁興磊高騰云劉賢梁棟付彤
畜牧獸醫學報 2024年9期



摘 要: 旨在通過遺傳多樣性和群體結構分析,評估南陽牛的保種效果。本研究基于30頭健康南陽牛的全基因組重測序數據,通過變異檢測獲得單核苷酸多態性(single nucleotide polymorphism, SNP)信息,綜合分析群體遺傳多樣性、群體結構、親緣關系以及連續純和片段(ROH)分布特征,對南陽牛保種群的保種效果進行全面評估。結果顯示:1)共檢測到高質量SNPs位點數目是25 929 389個;2)保種群遺傳多樣性豐富,核苷酸多樣性為0.002 9,多態性標記比例為0.888 7,最小等位基因頻率為0.186 1,期望雜合度為0.274 9,觀測雜合度為0.255 7;3)世代有效群體含量在1 000代前為2 834頭,在20代前為149頭,呈逐年下降趨勢;4)主成分分析結果顯示南陽牛保種群沒有明顯分層;5)個體間的遺傳距離介于0.80~0.89,平均遺傳距離為0.83±0.02;6)30頭南陽牛個體共分為7個家系,公牛可分為5個家系;7)共檢測到ROH片段數目是4 992個,平均每頭南陽牛的ROH片段長度為約74.41 Mb,總長度為2.18 Gb,基于ROH的平均近交系數為0.031,0.5 Mb以下的ROH片段占比最多為75.72%,2~4 Mb的ROH片段占比最少為0.16%。綜上所述,南陽牛保種群體遺傳多樣性豐富,沒有出現明顯分層,近交水平較低,但仍有個別個體近交較高。因此,未來的保種工作應加強選種和選配管理,以促進群體的可持續發展。
關鍵詞: 南陽牛;全基因組重測序;遺傳多樣性;群體結構;保種
中圖分類號: S823.2
文獻標志碼:A 文章編號: 0366-6964(2024)09-3876-11
Evaluation of the Conservation Effect in Nanyang Cattle Based on Resequencing Data
LIU" Siyu1, ZHANG" Man1, ZHANG" Yan1, WEI" Zhitong1, QI" Xinglei2, GAO" Tengyun1, LIU" Xian3, LIANG" Dong1*, FU" Tong1*
(1.College of Animal Science and Technology, Henan Agricultural University,
Zhengzhou 450046," China;2.Center of Animal Husbandry Technical Service in Biyang, Biyang 463700," China; 3.Henan Provincial Animal Husbandry Station, Zhengzhou 450008," China)
Abstract:" The study aimed to evaluate the conservation effect of Nanyang cattle by analyzing genetic diversity and population structure. In this study, based on the whole genome resequencing data of 30 healthy Nanyang cattle, single nucleotide polymorphism (SNP) information was obtained through mutation detection. The genetic diversity, population structure, phylogenetic relationship and runs of homozygosity (ROH) distribution characteristics were analyzed comprehensively to evaluate the conservation effect of Nanyang cattle. The results showed as follows: 1) The total number of high quality SNPs sites was 25 929 389; 2) The conservation population was rich in genetic diversity, nucleotide diversity was 0.002 9, polymorphism marker ratio was 0.888 7, minimum allele frequency was 0.186 1, expected heterozygosity was 0.274 9, observed heterozygosity was 0.255 7; 3) The effective population size was 2 834 before 1 000 generations and 149 before 20 generations, which showed a decreasing trend year by year; 4) The results of principal component analysis showed that there was no obvious stratification in Nanyang cattle conservation population; 5) The genetic distance between individuals ranged from 0.80 to 0.89, and the average genetic distance was 0.83±0.02; 6) The 30 Nanyang cattle were divided into 7 families, and the bulls were divided into 5 families; 7) A total of 4 992 ROH fragments were detected, the average length of ROH fragments per Nanyang cattle was about 74.41 Mb, the total length was 2.18 Gb, the average inbreeding coefficient based on ROH was 0.031, and the maximum proportion of ROH fragments below 0.5 Mb was 75.72%, the proportion of ROH fragments of 2-4 Mb was at least 0.16%. In summary, the genetic diversity of the Nanyang cattle conservation population is rich, there is no obvious stratification, the inbreeding level is low, but there are still some individuals with high inbreeding. Therefore, the future conservation work should strengthen the management of seed selection and mating, in order to promote the sustainable development of the population.
Key words: Nanyang cattle; whole genome resequencing; genetic diversity; population structure; breed conservation
*Corresponding authors:LIANG Dong, E-mail:554230517@qq.com; FU Tong, E-mail: futong2004@126.com
南陽牛產于河南省南陽市,是我國五大黃牛品種之一,具有體軀高大、結構勻稱、役肉兼用、耐粗飼、適應性強、性情溫順的特征。1988年將南陽牛收錄于《中國牛品種志》,2000年,農業部將南陽牛列入《國家畜禽品種保護名錄》,2006年列入《國家畜禽遺傳資源保護名錄》,標志著對其重要性的認可[1-2]。2002年,國家質量技術監督總局對南陽牛進行了原產地標記域名注冊,為南陽牛產業的快速發展奠定了基礎。南陽牛作為中國著名的地方優良品種之一,在畜牧業中占有重要地位[3]。但隨著社會上肉牛雜交改良力度的加大,南陽牛血統愈來愈混雜,純種南陽牛數量急劇下降,使得南陽牛保種及開發利用工作顯得尤為迫切[4]。南陽市采取了積極措施,設立了南陽市黃牛良種繁育場和南陽黃牛科技中心,核心群保持6個以上家系,并完善系譜和技術管理檔案。同時,河南省畜牧部門還通過與夏洛萊、皮埃蒙特、德國黃牛等品種進行改良,成功培育出了新品種夏南牛、皮南牛和德南牛,為南陽牛的保護和開發利用做出了重要貢獻[5-7]。因此,為了加強對南陽牛的保種和開發利用,不使地方肉牛資源出現缺失,南陽牛的保種任務已經迫在眉睫。
由于我國南陽牛保種工作起步較晚,技術、資金投入有限,缺乏系統的數據記錄以及實際生產中易出現記錄錯誤等原因,傳統基于系譜的遺傳結構分析容易受到缺失記錄和錯誤記錄的干擾,難以得出準確結論。作為一種高密度、多態性強且遺傳上穩定的技術,單核苷酸多態性(single nucleotide polymorphism, SNP)已成為研究畜禽遺傳變異的關鍵工具[8]。SNP分型主要通過SNP芯片、簡化基因組測序及全基因組重測序(whole genome sequencing, WGS)等方法,廣泛用于分析群體結構、進行全基因組選擇和評估種質資源[9-10]。WGS技術的成熟和成本降低促進了近年來對牛[11]、豬[12]、馬[13]、羊[14]、禽[15]等物種群體結構和遺傳多樣性研究的快速發展。近年來,已有多項研究利用WGS技術對南陽牛進行了全基因組重測序分析,為深入了解其血統組成和遺傳多樣性提供了重要見解[16-18]。但是,針對南陽牛保種效果進行全方面評估尚未見報道。
本研究聚焦南陽牛保種群體,采用全基因組重測序技術深入分析其群體結構和遺傳多樣性。通過評估保種效果,為南陽牛遺傳資源的有效保護和可持續開發利用提供科學的理論依據。本研究將深化對南陽牛遺傳特性和群體動態的理解,為其遺傳管理和持續利用提供實質性指導。
1 材料與方法
1.1 試驗材料
本試驗選取河南省南陽市黃牛良種繁育場30頭南陽牛(公牛12頭,母牛18頭)作為研究對象。所有試驗牛均在相同的設施條件和環境下的圈舍中飼養,并執行相同的飼養標準。在采樣時,這些牛均是健康狀態。所有樣本的耳組織被采集并存放在含95%酒精的2 mL凍存管中,-20 ℃保存。
1.2 試驗方法
1.2.1 DNA提取及測序
使用南京諾唯贊生物科技股份有限公司的組織基因組DNA提取試劑盒(DC112)提取南陽牛耳組織中的基因組DNA,并利用瓊脂糖凝膠電泳及紫外分光光度計評估其完整性、濃度與產量。使用DNBSEQ-T7平臺對每個個體的基因組DNA進行150 bp雙末端reads的測序。Trimomatic[19]軟件用于過濾fastq數據,生成clean reads。Clean reads通過BWA-MEM[20]算法與參考基因組(ARS-UCD1.3)進行比對。使用Picard[21]的MarkDuplicates模塊和GATK(v 3.8)[22]的IndelRealigner模塊分別去除PCR重復和重新比對indels。變異檢測采用UnifiedGenotyper模塊,過濾參數為:“QDlt;2.0, FSgt;60.0, 等。使用VCFtools(v 0.1.16)[23]將變異數據從VCF格式轉為Plink格式。質量控制中,本研究使用plink移除了最小等位基因頻率小于0.05、Hardy-Weinberg平衡P值小于10-6的SNP位點和缺失基因型大于10%的個體。
1.2.2 遺傳多樣性分析
本研究對SNP數據進行基因功能注釋所使用到的軟件是SnpEff[24]。主要是通過PLINK(v 1.90)[25]軟件計算南陽牛的最小等位基因頻率(minor allele frequency, MAF)、期望雜合度(expected heterozygosity, He)、觀測雜合度(observed heterozygosity, Ho)和多態性標記比例(proportion of polymorphic marker, PN),評估保種群的遺傳多樣性。VCFtools(v 0.1.16)軟件計算核苷酸多樣性(π),設置滑動窗口50 kb和步長20 kb。SNeP(v 1.1)[26]軟件基于連鎖不平衡計算歷史有效群體大小(Ne)。
1.2.3 南陽牛群體結構分析
本研究利用PLINK(v 1.90)軟件進行主成分分析(PCA),并用R語言的ggplot2包對前兩主成分進行可視化。通過PLINK計算遺傳距離,構建IBS距離矩陣和tassel[27]軟件構建G矩陣,R語言進行結果可視化。再次使用PLINK計算距離矩陣,通過R包“ape”、“phangorn”、“seqinr”建立NJ系統發育樹,利用iTOL(https://itol.embl.de/)網站進行可視化。
1.2.4 全基因組ROH統計分析
ROH檢測采用PLINK(v 1.90)[25]的-homozyg選項,以0.5~1 Mb、1~2 Mb、2~4 Mb、gt;4 Mb將其ROH分為4個等級并計算近交系數(FROH),通過R語言軟件包ggplot2可視化最終結果。
2 結 果
2.1 南陽牛群體遺傳多樣性分析
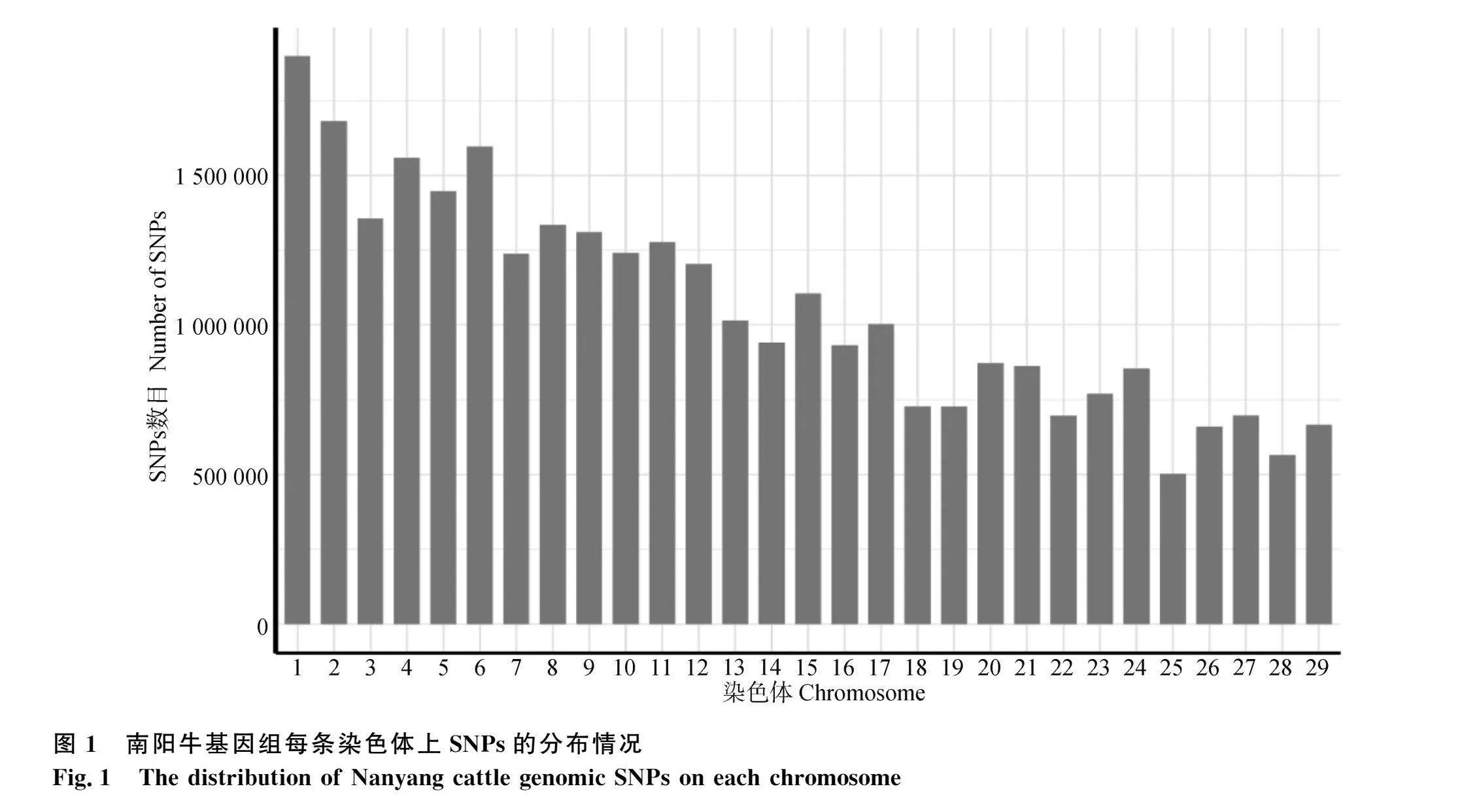
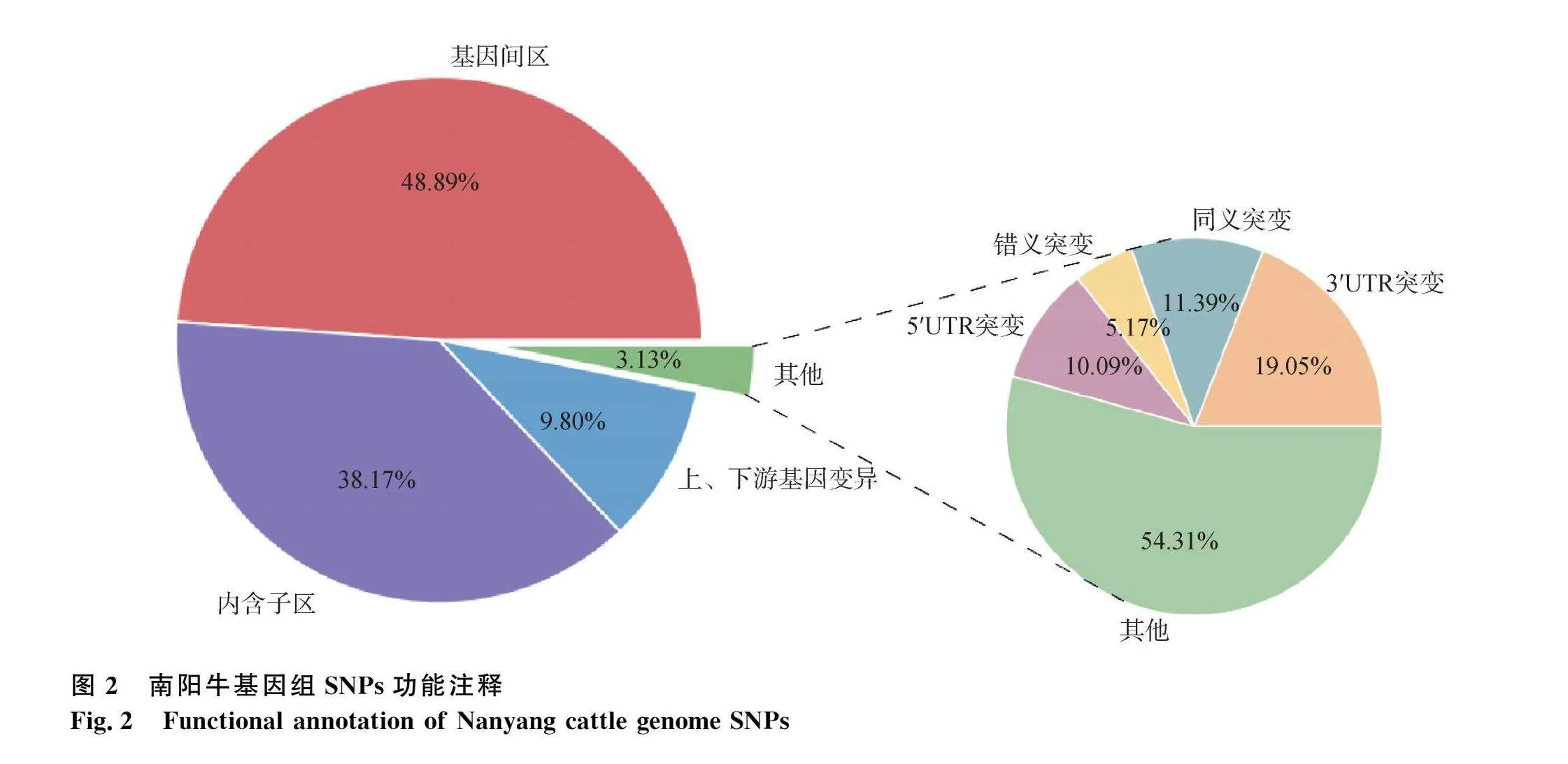
本研究對30頭南陽牛進行了全基因組重測序,平均測序深度達到12.65×。產生的clean reads有6 972 566 582個,其中平均99.87%的clean reads可以比對到參考基因組上。經過變異檢測和質量控制,30頭南陽牛共獲得25 929 389個高質量SNPs。在常染色體中,1號染色體上的SNP數目最多,包含1 897 811個SNPs;25號染色體上的SNP數目最少,包含502 704個SNPs(圖1)。基因功能注釋結果顯示(圖2),南陽牛全基因組中SNPs主要分布在基因間區(73 677 282個,占48.89%)以及內含子區(57 520 323個,占38.17%)。同義突變與錯義突變所占比例較少,分別包括462 843與209 938個SNPs。
南陽牛保種群遺傳多樣性的參數,包括最小等位基因頻率0.186 1,多態性標記比例0.888 7,期望雜合度0.274 9,觀測雜合度0.255 7,以及核苷酸多樣性為0.002 9,詳細信息見表1。觀測雜合度略低于期望雜合度,差異不顯著。
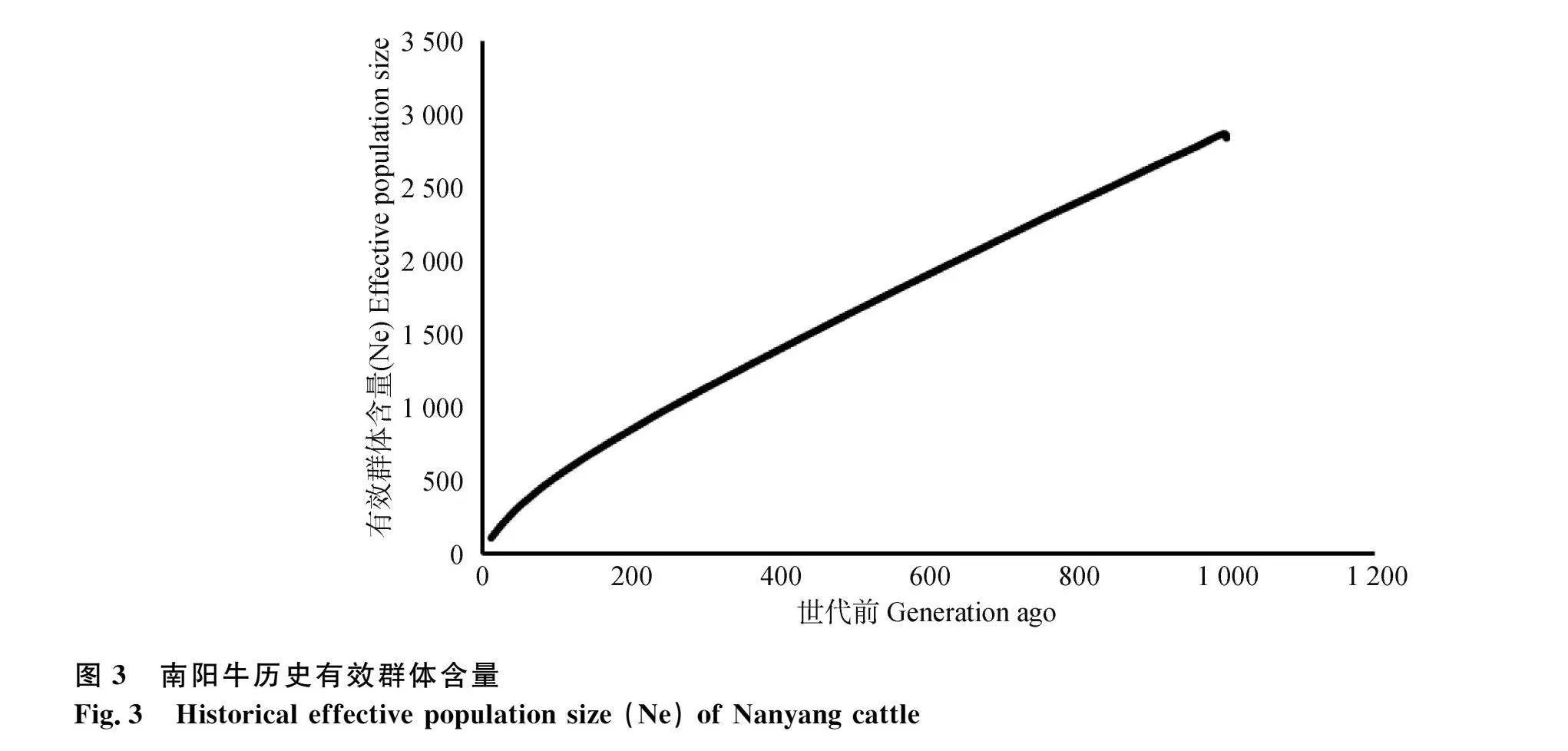
南陽牛保種群歷史有效群體大小的結果顯示,在1 000代前時,Ne為2 834頭;在150代前時,Ne為689頭;在20代前時,Ne為149頭(圖3)。這種變化趨勢說明南陽牛保種群的有效群體大小逐代減少。
2.2 南陽牛群體結構分析
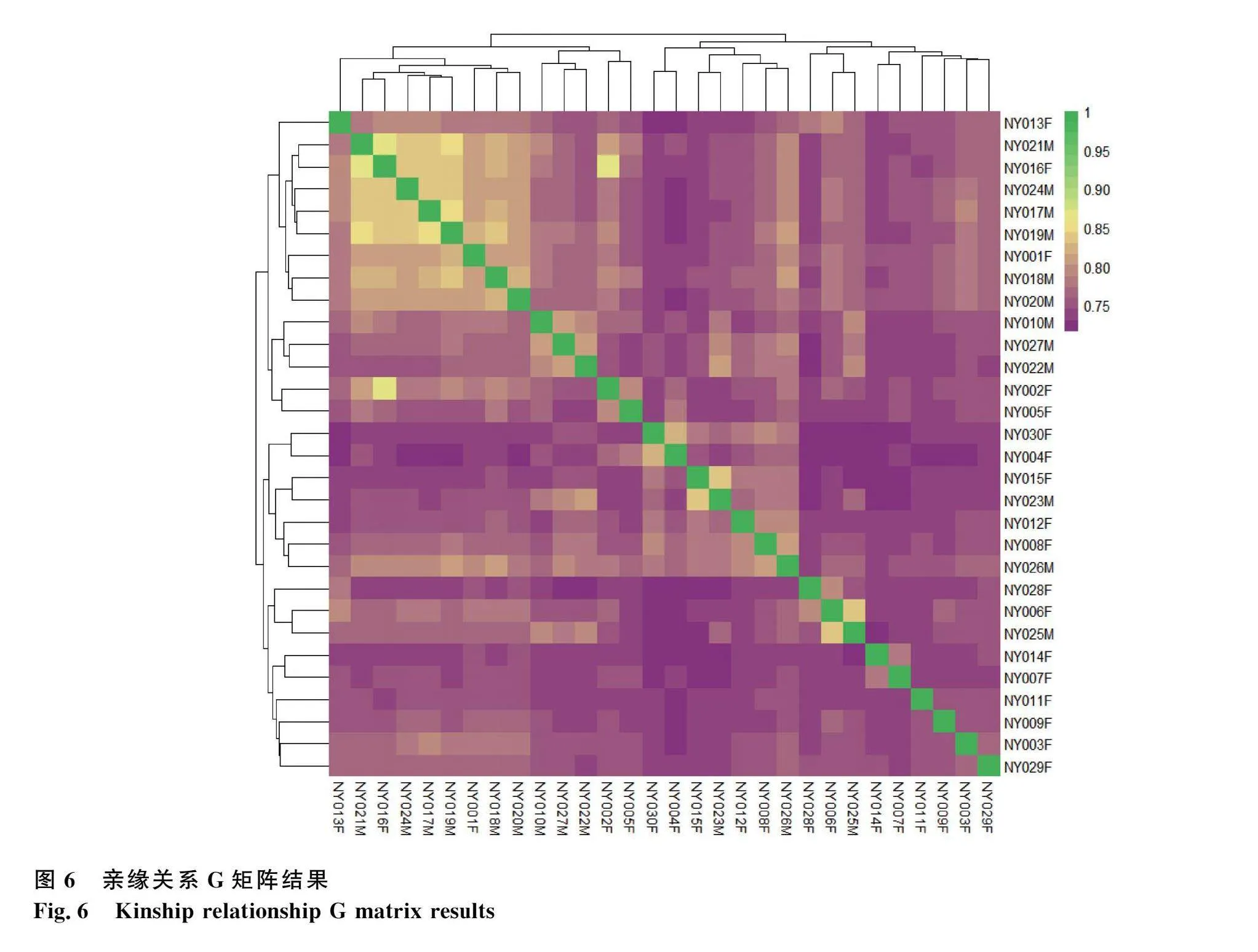
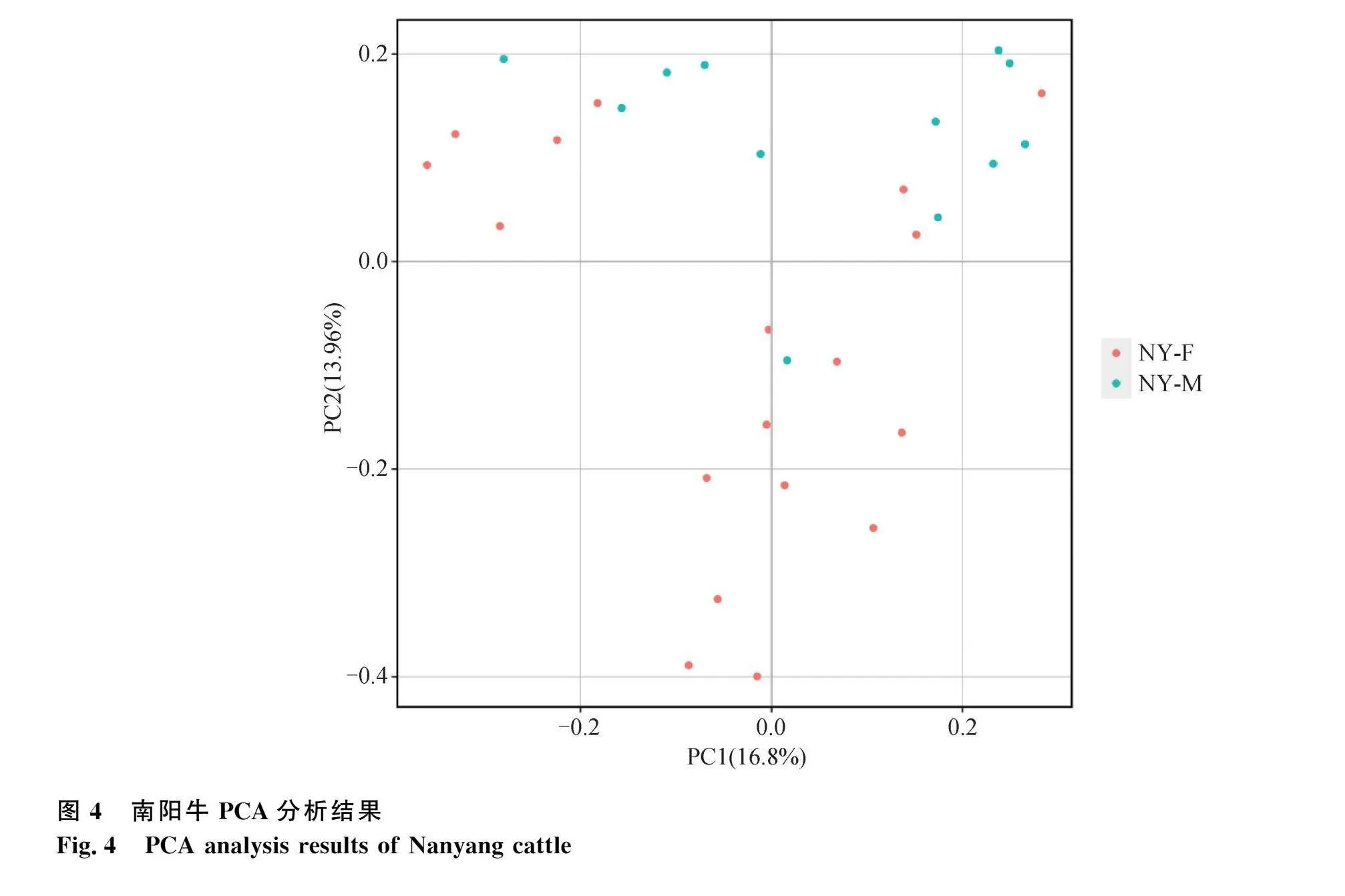
主成分分析結果顯示,主成分1解釋了16.8%的遺傳變異,主成分2解釋了13.96%的遺傳變異。保種群內部,12頭公牛(NY-M)分布均勻,未表現出明顯的聚集趨勢,見圖4所示。IBS的遺傳距離矩陣顯示,30頭南陽牛個體間的IBS遺傳距離范圍為0.80~0.89,平均值為0.83±0.02,見圖5。G矩陣揭示的親緣關系如圖6,通過顏色深淺表示,親緣關系系數接近1時顏色較淺,表明個體間關系較近。大多數南陽牛個體顯示中到低等級的親緣關系,僅少數表現出較高的親緣度。南陽牛保種群的"" 遺傳距離和親緣關系分析表明,個體間遺傳距離較大,近交程度低,但不排除潛在的近交風險。
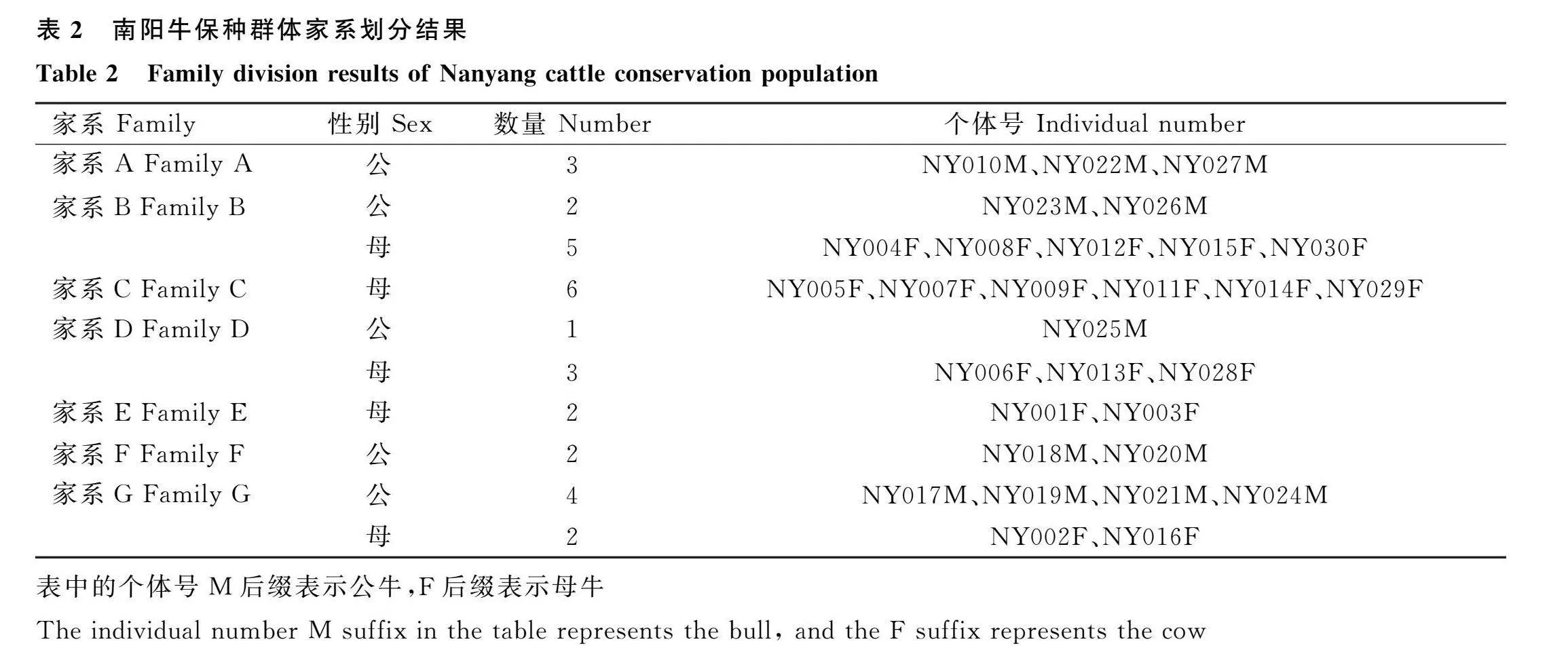
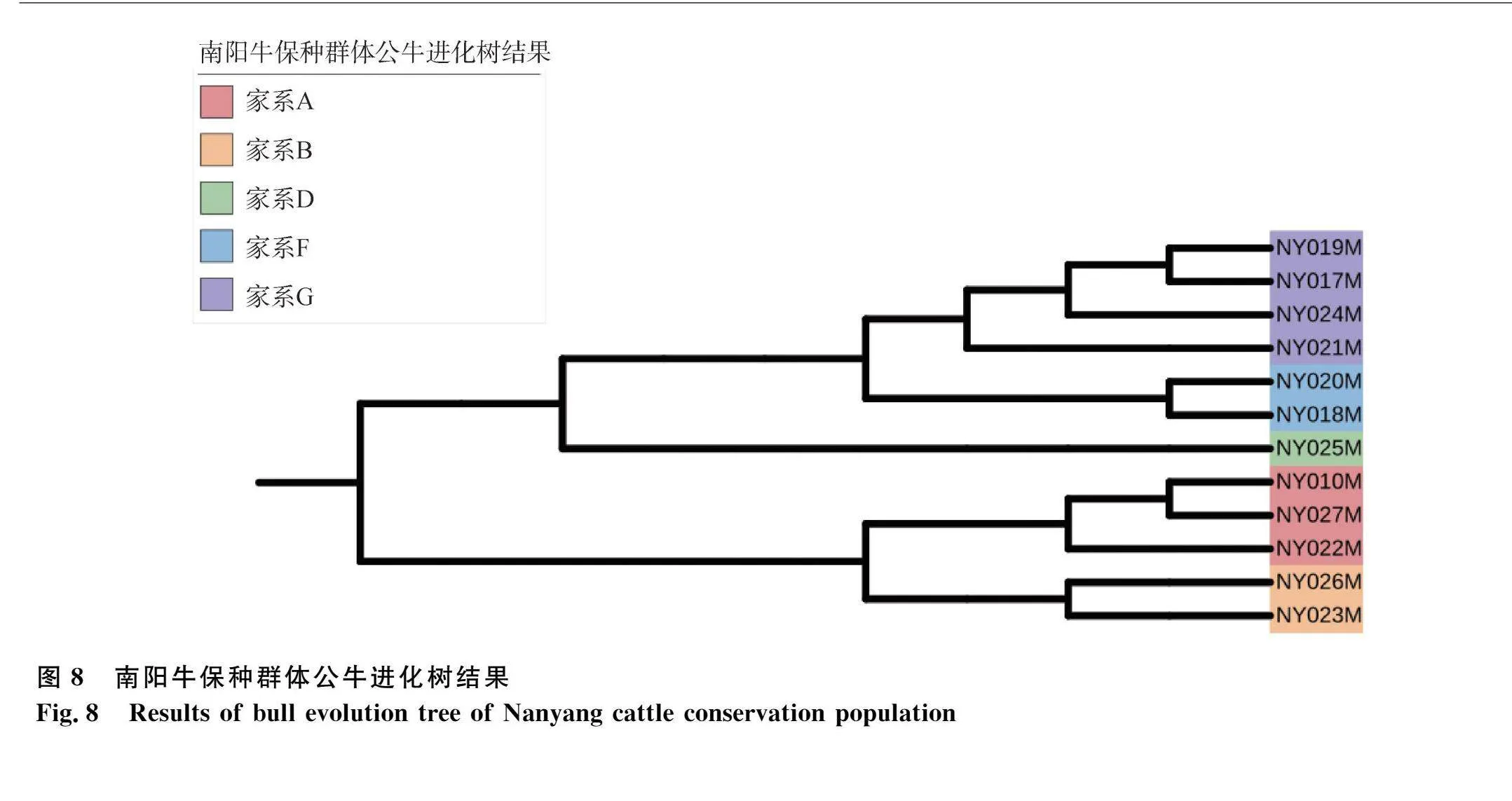
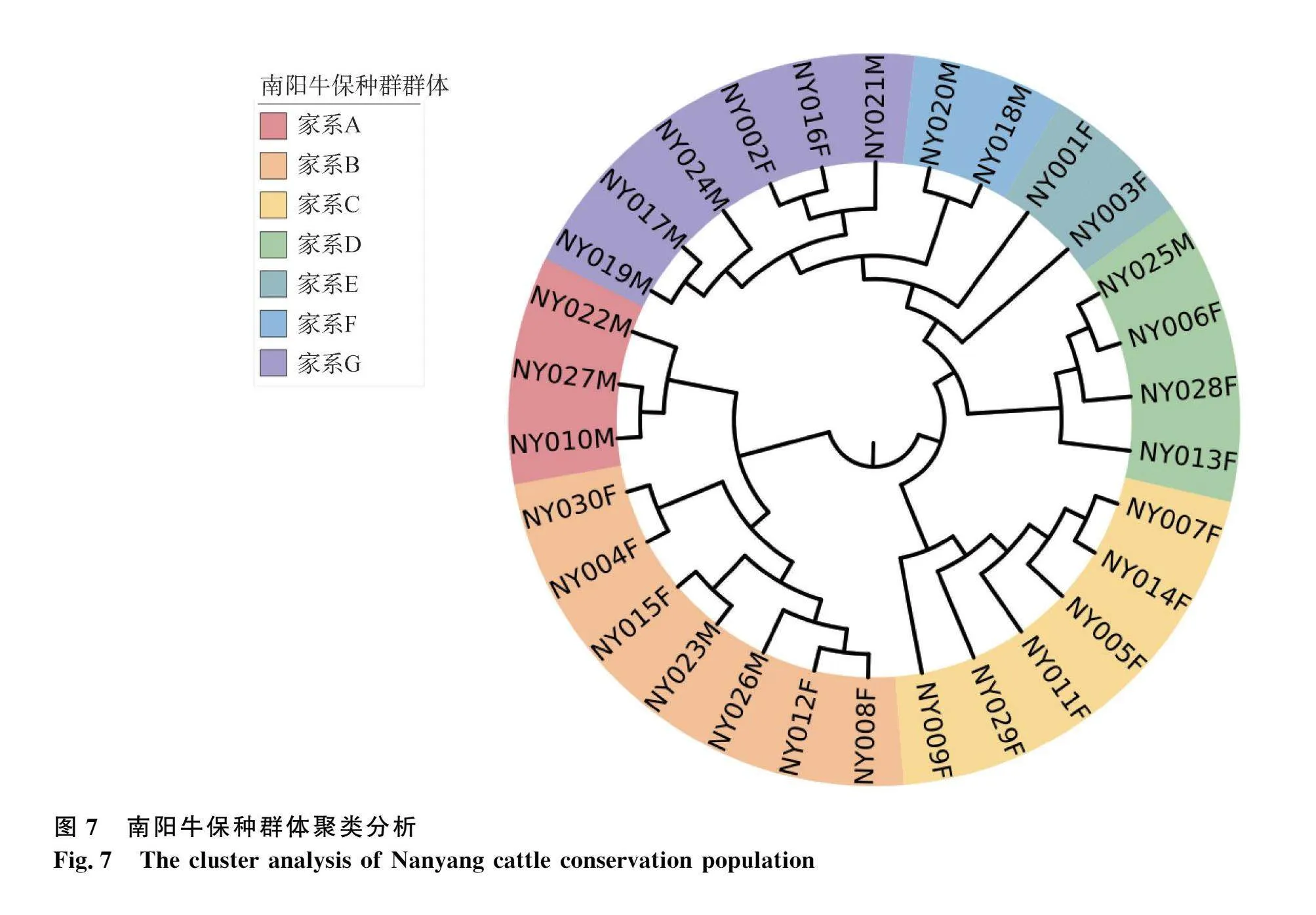
使用NJ法[28]為南陽牛保種群體構建了遺傳距離矩陣,并將親緣關系系數超過0.1的母牛歸入相應的公牛家系中,結果顯示30頭南陽牛可分為7個家系(圖7),而12頭公牛分為5個家系(圖8)。家系劃分如下:家系A包含3頭公牛;家系B包含2頭公牛和5頭母牛;家系C包含6頭母牛;家系D包含1頭公牛和3頭母牛;家系E包含2頭母牛;家系F包含2頭公牛;家系G包含4頭公牛和2頭母牛(表2)。
2.3 南陽牛群體基因組ROH及近交系數分析結果
30頭南陽牛中發現了4 992個ROH片段,平均每頭南陽牛約有166.4個ROH片段。所有ROH片段的總長度達2.18 Gb,平均每頭南陽牛的ROH片段長度約74.41 Mb。在所有染色體中,第7號染色體的ROH片段最多,共336個;而第25號染色體的最少,僅54個(圖9A)。對南陽牛ROH片段進行分類統計發現,0~0.5 Mb長度的片段最多,共3 780個,占總數的75.72%;而2~4 Mb長度的片段最少,共8個,僅占0.16%(圖9B)。南陽牛保種群的ROH長度和數量表明了其豐富的遺傳多樣性。30頭南陽牛ROH的近交系數介于0.012~0.058之間,平均值為0.031(圖9C)。
3 討 論
畜禽遺傳資源保護致力于維護遺傳多樣性,對國家畜產品供應和種業振興至關重要。中國是畜種業強國,培育了眾多牛品種,其中南陽牛作為杰出的地方品種,因其體型龐大、耐力強、適應粗飼、肉質鮮美及其特有的大理石紋理,在畜牧業發揮著關鍵作用,對肉牛產業和新品種培養極為重要。2000年,南陽牛被納入國家畜禽品種保護名錄,政府從此加強保護并開展了開發與選育。自1986年起,南陽牛數量不斷增加,一度達到約240萬頭。但隨著農業機械化的提高和外來品種的引入,自1997年起其數量開始逐年減少。為保護這一品質資源,南陽市建立了南陽市黃牛良種繁育場(原南陽黃牛場)和南陽黃牛科技中心(原南陽黃牛研究所),對南陽黃牛進行品種保護和改良利用。本研究通過對30頭南陽牛進行全基因組重測序,共獲取了25 929 389個高質量SNPs位點。利用這些數據評估了南陽牛的遺傳多樣性和群體結構,為保種工作提供了科學依據。
群體遺傳多樣性既映射了物種的進化軌跡,也展示了其對環境的適應性[29-32]。較高的遺傳多樣性通常預示著更強的環境適應力,對于促進遺傳資源的持續利用具有重要意義。Mei等[16]的研究表明,南陽牛群體的遺傳結構呈現出多樣性,并且在全球牛種中展現出獨特的遺傳特征;另一項研究由Zhang等[17]進行,發現南陽牛中存在著豐富的遺傳多樣性,并且呈現出一定程度的品種特異性;此外,Chen等[18]的研究進一步揭示了南陽牛的遺傳背景,指出其可能受到來自不同地區和品種的遺傳影響,表現出復雜的遺傳結構。這些研究結果表明,南陽牛的遺傳多樣性和血統組成受到多種因素的影響,包括地理環境、人工選擇和品種間交流等。根據已有研究,南陽牛是黃牛和瘤牛的雜交品種,其具有較高的遺傳多樣性[17]。未來的研究可以進一步探索南陽牛群體的遺傳背景和演化歷史,以更好地指導其保種工作和遺傳資源管理。
本研究對南陽牛群體的遺傳多樣性進行了深入分析。結果顯示,南陽牛群體的平均最小等位基因頻率為0.186 1,指示其遺傳多樣性較高。同時,58%的SNPs位點多態性進一步證實了這一點。南陽牛的平均觀測雜合度為0.255 7,略低于期望值,暗示可能存在輕微的選擇壓力和近交現象。通過連鎖不平衡估計,20代前南陽牛的有效群體規模約為149頭,顯示下降趨勢,這說明南陽牛群體仍保持較高的遺傳多樣性,但仍然還存在遺傳多樣性喪失的風險。根據本研究結果,未來遺傳管理和持續利用工作可以考慮以下改進措施:制定科學的繁殖計劃,避免近親繁殖,選擇遺傳多樣性高的個體進行配種。收集南陽牛的遺傳信息,為種群管理提供數據支持。在保持遺傳多樣性的同時,通過人工選擇引導種群進化。提高養殖戶對遺傳多樣性保護和繁殖管理的意識和能力,確保管理措施的有效實施。
為防止外來品種干擾,保種場多采用內群繁育,使得研究保種群體結構對于其可持續發展極為重要[33-34]。主成分分析顯示,30頭南陽牛沒有明顯分層,遺傳距離分析表明個體間親緣關系較遠,暗示近交程度較低。聚類分析將30頭南陽牛分為7個家系,顯示了家系間的個體數量差異。未來保種應關注公牛分布,優化配種策略以保證家系平衡。ROH產生于親代將同源單倍型完整傳遞給后代。長ROH片段表明近期的近交歷史,而短ROH源自遠祖。研究發現,30頭南陽牛中共有4" 992個ROH片段,總長2.18 Gb。其中,0~0.5 Mb的ROH片段最多,而2~4 Mb的最少。這說明南陽牛近期無顯著近交。平均近交系數為0.031,反映了南陽牛整體近交水平較低。為保持低近交水平,選配時應注意高近交個體,引入新血統或遠親家系的公牛進行配種可以有效降低其后代的近交系數。也可以通過分子標記技術識別近交個體,從而在選種和配種時予以排除。
4 結 論
本研究利用全基因組重測序數據分析了30頭南陽牛的遺傳多樣性和群體結構。發現南陽牛保種群體遺傳多樣性較高,群體結構未顯著分層。通過聚類分析,將這些牛分為7個家系,揭示了它們之間的親緣關系。雖然大部分南陽牛近交水平較低,但仍有個別個體近交較高。因此,未來的保種工作應加強選種和配種管理,以促進群體的可持續發展。
參考文獻(References):
[1] XIA X,QU K,ZHANG G,et al.Comprehensive analysis of the mitochondrial DNA diversity in Chinese cattle[J].Anim Genet,2019,50(1):70-73.
[2] ZHANG Y,WEI Z T,ZHANG M, et al.Population structure and selection signal analysis of Nanyang cattle based on whole-genome sequencing data[J].Genes,2024,15(3): 351.
[3] XIA X,YAO Y,LI C,et al.Genetic diversity of Chinese cattle revealed by Y-SNP and Y-STR markers[J].Anim Genet,2019,50(1): 64-69.
[4] 陳希鵑,肖喜東,吳明安.南陽黃牛的保種、改良與種公牛飼養管理[J].養殖與飼料,2020(3):47-48.
CHEN X J,XIAO X D,WU M A.Breeding,improvement and breeding management of Nanyang yellow cattle[J].Breeding and Feed,2020(3):47-48.(in Chinese)
[5] 杜書增,王冠立,王玉海,等.南陽牛肉用選育改良及其產業化開發[J].中國牛業科學,2016,42(2):63-66.
DU S Z,WANG G L,WANG Y H,et al.Breeding and improvement of Nanyang cattle and its beed industrialization[J].China Cattle Science,2016,42(2):63-66.(in Chinese)
[6] 歲豐軍.南陽牛保種育種現狀[C]//《第八屆中國牛業發展大會》論文集.昌吉:中國畜牧業協會,2013:248-251.
SUI F J.Current situation of Nanyang cattle breeding[C]//Proceedings of the 8th China Cattle Industry Development Conference. Changji:China Animal Husbandry Association,2013:248-251.(in Chinese)
[7] 茹寶瑞,高騰云.關于南陽牛育種與生產的一些建議[C]//《第六屆中國牛業發展大會》論文集.重慶:中國畜牧業協會牛業分會,2011:310-313.
RU B R,GAO T Y.Some suggestions on breeding and production of Nanyang cattle[C]//Proceedings of the 6th China Cattle Industry Development Conference.Chongqing:China Animal Husbandry Association Cattle Industry Branch,2011:310-313.(in Chinese)
[8] LIANG D,ZHAO P J,SI J F,et al.Genomic analysis revealed a convergent evolution of LINE-1 in coat color:a case study in water buffaloes (Bubalus bubalis)[J].Mol Biol Evol,2021,38(3):1122-1136.
[9] KARIMI K,KOSHKOIYEH A E,FOZI M A,et al.Prioritization for conservation of Iranian native cattle breeds based on genome-wide SNP data[J].Conserv Genet,2016,17(1):77-89.
[10] 劉繼強,郝曉東,武麗娜,等.全基因組SNP分型技術在畜禽遺傳育種研究中的應用[J].畜牧獸醫學報,2022, 53(12): 4123-4137.
LIU J Q,HAO X D,WU L N,et al.Application of whole genome SNP genotyping technology in livestock and poultry genetics and breeding[J].Acta Veterinaria et Zootechnica Sinica,2022,53(12):4123-4137.(in Chinese)
[11] HU M Y,SHI L L,YI W F,et al.Identification of genomic diversity and selection signatures in Luxi cattle using whole-genome sequencing data[J].Anim Biosci,2024,37(3):461-470.
[12] 龍 熙,柴 捷,潘紅梅,等.基于SNP芯片的盆周山地豬群體選擇信號分析[J].中國畜牧雜志,2023,59(12):136-140.
LONG X,CHAI J,PAN H M,et al.Signal analysis of population selection of mountainous pigs in pelvic region based on SNP chip[J].Chinese Journal of Animal Science,2023,59(12):136-140.(in Chinese)
[13] JAGANNATHAN V,GERBER V,RIEDER S,et al.Comprehensive characterization of horse genome variation by whole-genome sequencing of 88 horses[J].Anim Genet,2019,50(1):74-77.
[14] 史露露,胡明月,賴偉寧,等.基于50K SNP芯片的夏洛來羊遺傳結構及選擇信號分析[J].黑龍江畜牧獸醫,2023(6): 49-53,59,139-140.
SHI L L,HU M Y,LAI W N,et al.Genetic structure and selection signal analysis of Charolais sheep based on 50K SNP chip[J].Heilongjiang Animal Science and Veterinary Medicine,2023(6):49-53,59,139-140.(in Chinese)
[15] GUO Y,RUBIN G J,R NNEBURG T,et al.Whole-genome selective sweep analyses identifies the region and candidate gene associated with white earlobe color in Mediterranean chickens[J].Poult Sci,2024,103(1):103232.
[16] MEI C G,WANG H C,LIAO Q J,et al.Genetic architecture and selection of Chinese cattle revealed by whole genome resequencing[J].Mol Biol Evol,2018,35(3):688-699.
[17] ZHANG S J,YAO Z,LI X M,et al.Assessing genomic diversity and signatures of selection in Pinan cattle using whole-genome sequencing data[J].BMC Genomics,2022,23(1):460.
[18] CHEN N B,CAI Y D,CHEN Q M,et al.Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia[J].Nat Commun,2018,9(1):2337.
[19] BOLGER A M,LOHSE M,USADEL B.Trimmomatic:a flexible trimmer for Illumina sequence data[J].Bioinformatics, 2014,30(15):2114-2120.
[20] LI H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM[J]. arXiv preprint,2013,arXiv: 1303.3997.
[21] EBBERT M T W,WADSWORTH M E,STALEY L A,et al.Evaluating the necessity of PCR duplicate removal from next-generation sequencing data and a comparison of approaches[J].BMC Bioinformatics,2016,7:239.
[22] MCKENNA A,HANNA M,BANKS E,et al.The genome analysis toolkit:a MapReduce framework for analyzing next-generation DNA sequencing data[J].Genome Res,2010,20(9):1297-1303.
[23] DANECEK P,AUTON A,ABECASIS G,et al.The variant call format and VCFtools[J].Bioinformatics,2011,27(15):2156-2158.
[24] CINGOLANI P.Variant annotation and functional prediction:SnpEff[J].Methods Mol Biol,2022,2493:289-314.
[25] PURCELL S,NEALE B,TODD-BROWN K,et al.PLINK:a tool set for whole-genome association and population-based linkage analyses[J].Am J Hum Genet,2007,81(3):559-575.
[26] BARBATO M,OROZCO-TERWENGEL P,TAPIO M,et al.SNeP:a tool to estimate trends in recent effective population size trajectories using genome-wide SNP data[J].Front Genet,2015,6:109.
[27] BRADBURY P J,ZHANG Z W,KROON D E,et al.TASSEL:software for association mapping of complex traits in diverse samples[J].Bioinformatics,2007,23(19):2633-2635.
[28] WANG Y,DONG R L,LI X,et al.Analysis of the genetic diversity and family structure of the Licha Black pig population on Jiaodong Peninsula,Shandong Province,China[J].Animals (Basel),2022,12(8):1045.
[29] EDWARDS C E,TESSIER B C,SWIFT J F,et al.Conservation genetics of the threatened plant species Physaria filiformis (Missouri bladderpod) reveals strong genetic structure and a possible cryptic species[J].PLoS One,2021,16(3):e0247586.
[30] 劉晨龍,盧 丹,周泉勇,等.利用高密度SNP芯片分析杭豬的群體遺傳結構[J].畜牧獸醫學報,2022,53(8):2502-2513.
LIU C L,LU D,ZHOU Q Y,et al.Analysis of population genetic structure of hang pigs by high density SNP chip[J].Acta Veterinaria et Zootechnica Sinica,2022,53(8):2502-2513.(in Chinese)
[31] 陳國宏,季從亮,王敏強.12個中國地方雞種群體遺傳結構及遺傳多樣性分析[J].中國畜牧獸醫文摘,2006(5):32.
CHEN G H,JI C L,WANG M Q.Analysis of population genetic structure and genetic diversity of 12 local chicken breeds in China[J].Chinese Journal of Animal Science and Veterinary Abstracts,2006(5):32.(in Chinese)
[32] 李隱俠,牙生江·那斯爾,賽里克·都曼,等.SNP芯片評估柯爾克孜羊群體遺傳多樣性和遺傳結構[J].畜牧獸醫學報,2023,54(2):572-583.
LI Y X,YA SHENGJIANG·NASIER,SAI LIKE·DUMAN,et al.Evaluation of genetic diversity and genetic structure in Kirgiz sheep population based on SNPs chip[J].Acta Veterinaria et Zootechnica Sinica,2023,54(2):572-583.(in Chinese)
[33] 馬克巖,韓金濤,白雅琴,等.基于簡化基因組測序的永登七山羊遺傳多樣性分析[J].畜牧獸醫學報,2023,54(5):1939-1950.
MA K Y,HAN J T,BAI Y Q,et al.Genetic diversity analysis of Yongdeng Qishan sheep based on specific-locus amplified fragment sequencing[J].Acta Veterinaria et Zootechnica Sinica,2023,54(5):1939-1950.(in Chinese)
[34] 馬 鈞,樊安平,王武生,等.全基因組重測序解析秦川牛保種群遺傳多樣性和遺傳結構[J].遺傳,2023,45(7):602-616.
MA J,FAN A P,WANG W S,et al.Analysis of genetic diversity and genetic structure of Qinchuan cattle conservation population using whole-genome resequencing[J].Hereditas,2023,45(7):602-616.(in Chinese)
(編輯 郭云雁)
