固相萃取凈化/GC-MS法測定水/土樣品中α-松油醇含量
2024-11-09 00:00:00吳鳳區嘉鋮李彥趙海明蔡全英向壘莫測輝
農業環境科學學報 2024年9期

摘要:采用固相萃取凈化結合GC-MS法分別建立了測定水/土樣品中典型有機浮選藥劑(α-松油醇)的方法。采用DB-5 MS進行色譜分離,電子轟擊(EI)-選擇離子檢測(SIM)和基質標線進行定量分析。水/土樣品均采用二氯甲烷進行萃取,水樣萃取時,先將其pH值和鹽度調整為7.5和0.5%,之后采用二氯甲烷進行3次萃取;土壤萃取時采用二氯甲烷進行2次振蕩萃取,萃取液旋轉蒸發近干后用1 mL甲醇定容為萃取質。水、土樣品萃取質均采用Cl8小柱進行固相萃取凈化。在上述最優萃取條件下,目標化合物在0.1-5mg·L-1濃度范圍內線性關系良好(R2≥0.998);其在水/土樣品中的檢出限(LOD)分別為0.027-0.041 mg·L-1(水樣)和0.009-0.054 mg·kg-1(土樣),在實際樣品中不同濃度(1、2、5 mg·L-1或1、2、5 mg·kg-1)加標回收率分別為91.5%-112%[相對標準偏差(RSD)< 15%,水樣]和67%-114%(RSD<15%,土樣)。
關鍵詞:有機選冶藥劑;選礦廢水;土壤;松油醇;GC-MS
中圖分類號:X830.2;0657.63 文獻標志碼:A 文章編號:1672-2043(2024)09-2183-08 doi:10.11654/jaes.2024-0638
礦業作為國民經濟的基石,為我國工業化進程提供了堅實的物質基礎,扮演著不可或缺的角色。然而,礦業活動會導致重金屬和選冶藥劑被釋放進入環境造成嚴重污染。值得注意的是,以往的研究主要關注重金屬污染而忽略了選冶藥劑污染。事實上,礦業活動大量使用選冶藥劑,其全球年使用量達400萬t以上,其中我國選冶藥劑使用規模最大,占全球市場份額的60%以上,每年產生的有機浮選廢水量超過12億m3。有機浮選藥劑在浮選廢水中的殘留量通常能達到其使用量的50%以上,它們通常具有中/高毒性,進入環境后可造成嚴重的負面生態環境效應,如低濃度條件(mg·L-1水平)即可造成水生生物生長和繁殖的抑制,甚至導致其死亡。
松油醇(Terpineol,CIOH180)也稱為松脂醇(俗稱“2號油”),由松節油作為原料通過水合反應并經脫水蒸餾獲得,為環狀萜烯結構,分子量為154,包含3種同分異構體,即α-松油醇、β-松油醇和γ-松油醇,通常為淺黃色透明液體,有刺激性氣味,可微溶于水,易溶于有機溶劑,具有弱極性。由于具有優異穩定的起泡性能,松油醇常被作為起泡劑用于多種金屬(如鐵、銅、鉛和鋅礦石等)和非金屬(煤、滑石、石墨等)的浮選工藝。一般而言,松油醇起泡劑的主要成分是α-松油醇。松油醇屬于強毒性浮選藥劑,其對魚類的48 h LC50(半致死濃度)為8.74 mg·L-1,對橢圓蘿卜螺和大臍圓扁螺的96 h LC50分別為5.79mg·L-1和11.86 mg·L-1,其毒性分別為C3-C10醇類(10號油)和芐基多縮乙二醇類(甘芐油)浮選藥劑毒性的6.7倍和10.1倍。因此,開展松油醇化合物污染特征、環境行為等方面的研究十分緊迫。然而,目前相關研究尚鮮見報道,這與現有分析方法不完善有關。
目前有關松油醇的分析方法主要針對水樣或松油醇工業品(如精油等),包括滴定法、分光光度計法、液相色譜法和氣相色譜法等。其中滴定法需將松油醇氧化為碘化松油醇,方法步驟繁瑣、滴定終點不易觀察;分光光度計法靈敏度差,測定效果易受樣品基質成分干擾;色譜法有效提升了目標化合物檢測靈敏度,但定性效果相對較差,同樣易受樣品基質成分干擾。因此,亟待建立針對松油醇色譜—質譜聯用測定方法,并通過優化實驗解決樣品基質成分干擾的問題。另外,目前主要針對礦區廢水中重金屬開展治理,采用的方法包括化學沉淀法、吸附法、離子交換法等,但這些方法對廢水中的選冶有機藥劑去除效果有限,通常導致使用量50%以上的選冶有機藥劑隨浮選廢水排放進入環境,進而通過灌溉、溢流等方式進入土壤造成污染。因此,土壤中松油醇的可靠分析方法也有必要建立。基于上述原因,本研究建立了固相萃取凈化和氣相色譜/質譜聯用(GC-MS)分別測定不同性質水樣和土壤中α-松油醇的分析方法,并成功用于實際環境樣品的測定,獲得滿意的靈敏度、回收率和精密度。
1 材料與方法
1.1 實驗材料與儀器
α-松油醇(98%)購自Aladdin上海有限公司,分子量為154。甲醇、二氯甲烷和三氯甲烷均為色譜純,購自Sigma-Aldrich。腐植酸(FA≥90%)購自上海麥克林生化科技股份有限公司。其余試劑如氯化鈉(NaCl)、鹽酸(HCl)、氫氧化鈉(NaOH)等均為分析純,購自廣州化學試劑廠。實驗用水為超純水,由Unique-R20水機制備,其電阻率為18 MΩ·cm。
實驗儀器包括Model2010-Model QP2010氣相色譜-質譜聯用儀(日本島津公司)、RE-2000施蒸儀(中國上海亞榮)、SK15高速冷凍離心機、S1-234萬分之一電子分析天平(Denver Instrument Germany)、超純水儀(中國廈門科學儀器有限公司)、YD-1H筆試鹽度計。
1.2 實驗方法
標準溶液配制:準確移取0.1 g松油醇以色譜純甲醇定容于100 mL容量瓶,獲得目標化合物標準儲備液(1 000 mg·L-1),密封,于4℃保存備用。目標化合物標準曲線工作液(0.01-5 mg·L-1)由儲備液稀釋獲得,現用現配。
水樣萃取:取50 mL水樣過濾后,用1.0 mol·L-1HCl調節pH值為7.5,用5.0 mol·L-1 NaCl調節水樣鹽度為0.5%(m/V),置于125 mL分液漏斗中,用二氯甲烷萃取3次,每次用量為5.0 mL。合并萃取液于雞心瓶中,用選裝蒸發儀濃縮至近干,用1 mL甲醇定容,過C18固相萃取小柱凈化,收集濾液過0.22 μm尼龍PA濾膜(Pall Corporation),4℃保存備測。
土樣萃取:準確稱取經過凍干的1.0 g土壤置于50 mL聚丙烯離心管中,用二氯甲烷作為萃取劑萃取兩次,每次用量為5.0 mL,振蕩8 min(2 500 r·min-1)。萃取后,離心(8 000 r·min-1,10 min)獲得萃取上清液。合并兩次萃取上清液旋蒸至近干,用1.0 mL甲醇定容,過C18固相萃取小柱凈化,收集濾液過0.22 μm尼龍PA濾膜,4℃保存備測。
1.3 色譜-質譜條件
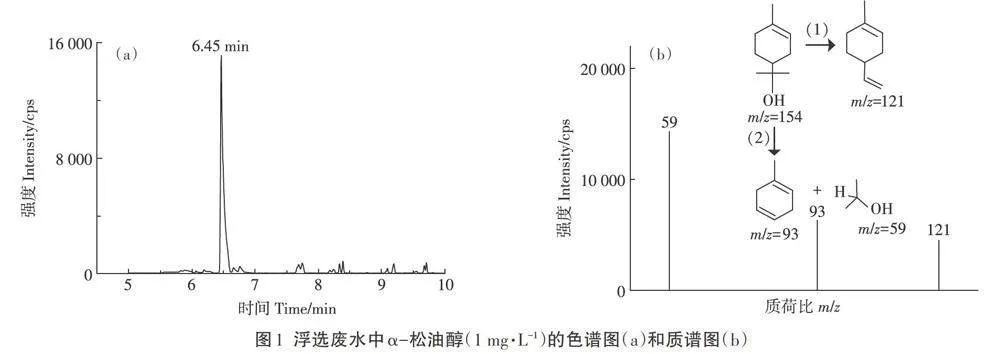
采用DB-5 MS進行色譜柱分離(30 m×0.25 mm×0.1 μm)。載氣為高純氮氣(≥99.999%),總流量50mL·min-1,柱流量0.95 mL.min-1,線速度35.8 cm·min-1,吹掃速度3 mL·min-1。以電子轟擊(EI)-選擇離子模式(SIM)進行定量檢測,其中定量離子為m/z=59,定性離子為m/z=93和m/z=121。進樣量1μL,進樣口溫度260℃,不分流進樣。離子源溫度180℃,接口溫度260℃,檢測電壓1.1 kV。梯度升溫程序:初始溫度70℃,以10℃·min-1速度升高至120℃,再以20℃·min-1升溫至200℃,保留1 min,總運行時長為10 min,目標化合物保留時間為6.45 min。
2 結果與討論
2.1 色譜-質譜條件優化
比較了3種常用于揮發性有機化合物色譜且具有相同規格(30 m×0.25 mm×0.25 μm)不同固定相的色譜柱(DB-5 MS、Rtx-5 MS、SH-Rxi-5 SilMS)對目標化合物(1 mg·L-1)的出峰效果。結果表明,DB-5MS的目標化合物的出峰效果峰形好、無峰拖尾或分叉現象,且信號響應最強,在空白浮選廢水中所受到的雜質干擾小、選擇性好(圖1a)。DB-5 MS、Rtx-5MS、SH-Rxi-5 SilMS色譜柱的固定相分別為5%苯基+95%甲基聚硅氧烷、5%二苯基+95%二甲基聚硅氧烷、交聯1,4-二(二甲基)苯基甲基聚硅氧烷。3種色譜柱均為弱極性色譜柱,可適用于弱極性化合物如多環芳烴、鄰苯二甲酸酯、有機醇類的分析,其中DB-5 MS色譜柱對α-松油醇出峰效果最好(色譜保留時間為6.45 min),因此被選擇用于后續優化實驗。
采用EI源-全掃描離子模式分析α-松油醇(1mg·L-1)的特征離子。如圖1b所示,在EI源電子轟擊條件下,a-松油醇形成3個特征離子,即準離子分子[(CH3)2-COH]+(m/z=59)、[C6H7—CH3]+(m/z=93)和[CH3—C6H8—C=CH2]+(m/z=121)(圖1b)。這一結果表明,EI源作用下α-松油醇發生兩類斷裂:一是脫去1個CH3和—OH形成[CH3—C6H8—C=CH2]+分子離子(m/z=121);二是脫去CH3—C(OH)—CH3形成[(CH3)2COH]+分子離子(m/z=59)和[C6H7—CH3]+分子離子(m/z=93)。3個特征離子中[(CH3)2—COH]+(m/z=59)豐度最高且響應靈敏,因此選為定量離子,另2個準分子離子則作為定性離子,用于后續的SIM模式分析。
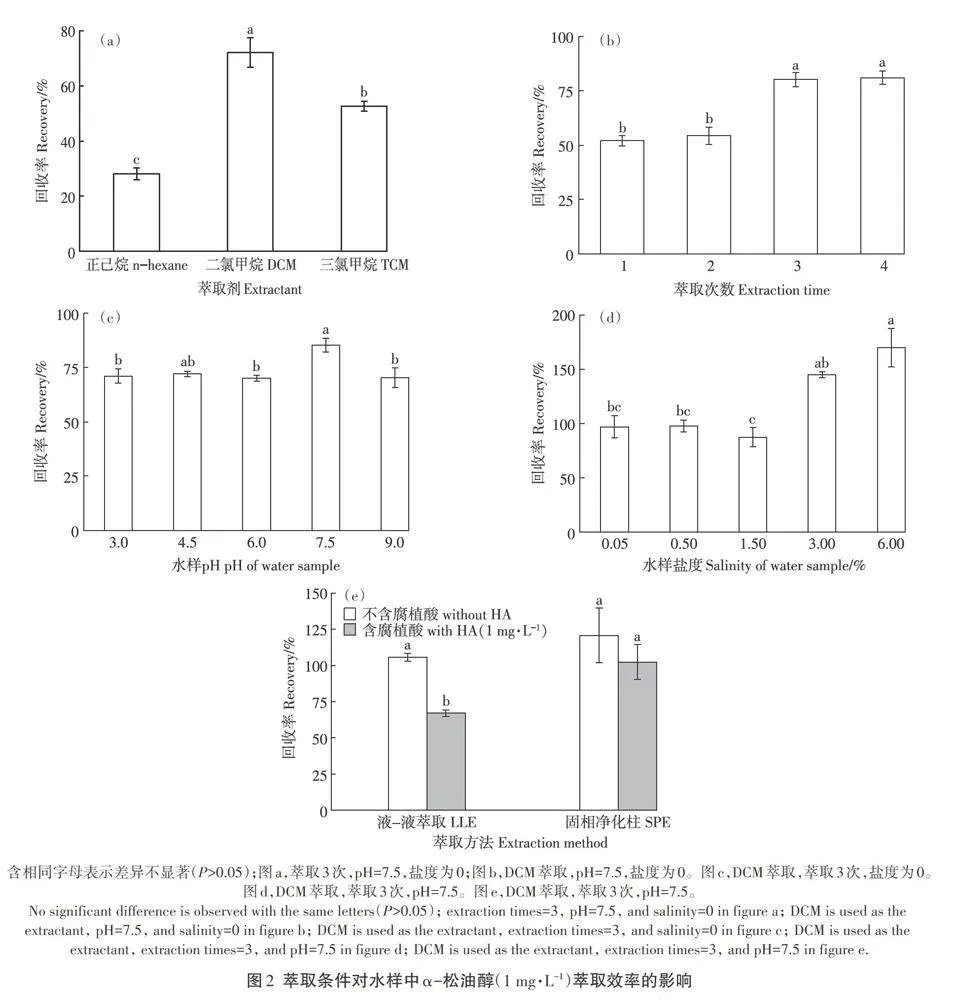
2.2 水樣萃取條件優化
液-液萃取要求所用萃取劑與水互不相溶且對待測化合物有較強溶解力和選擇性。本研究考察了不同萃取劑(二氯甲烷、三氯甲烷和正己烷)對目標化合物萃取效率的影響。結果表明,二氯甲烷對α-松油醇(1 mg·L-1)的萃取回收率最高(72%),而正己烷(28%)和三氯甲烷(52%)的萃取效率均低于60%(圖2a)。這一結果可能與目標化合物及不同萃取劑的極性匹配性有關。所用3種萃取劑極性大小關系為三氯甲烷(4.4)>二氯甲烷(1.6)>正己烷(0)。因此,根據“相似相溶”的原則,具有中等偏弱極性的二氯甲烷較另外兩種萃取劑更適合萃取同樣具有中等極性的目標化合物α-松油醇,因此選擇該溶劑進一步開展后續實驗。
以二氯甲烷作為萃取劑進一步考察了不同萃取次數(1-4次)對目標化合物(1 mg·L-1)萃取效率的影響。結果表明,萃取1次和2次時,α-松油醇的回收率低于60%,至萃取3次時其回收率提升至約80%,但4次萃取并未顯著提升萃取回收率,因此選擇萃取次數為3次(圖2b)。考慮到pH值和鹽度也是影響萃取效率的重要因素,本研究進一步考察了水樣不同pH值(3-9)和鹽度(0-6%)對目標化合物萃取效率的影響。結果表明,中性pH值條件下,目標化合物回收率約為85%,優于酸性條件(pH=3-6,回收率約為70%)和堿性條件(pH=9,回收率約為70%,圖2c),故選擇調節水樣pH為7.5。對于鹽度而言,提升水樣鹽度為0.05%和0.50%時,目標化合物回收率提升至約96%,較未添加處理提升約10%(圖2d)。這是由于鹽度的適度增加有助于促進萃取劑二氯甲烷與水溶液的有效分離,進而降低萃取劑在液-液萃取過程中因人工振搖而產生的乳化現象,從而減少目標化合物的損失。然而,進一步提升鹽度至3%以上時,目標化合物回收率超過150%,這可能是由于高鹽度影響了目標化合物的離子化效率,導致HP-MS/MS分析時的基質增強效應,從而造成目標化合物的異常高回收率。另外,實驗中觀察到水樣鹽度為0.50%時,萃取劑(二氯甲烷)乳化作用最弱,且目標化合物回收率標準偏差低于水樣鹽度,為0.05%,因此選擇調節水樣鹽度為0.50%。
環境水樣中通常存在有機質成分尤其是腐殖質等,這些成分的存在會對目標化合物的萃取檢測造成影響。因此根據環境水樣中有機質成分的濃度范圍(0.3-4.0 mg·L-1),在上述優化實驗的基礎上,以5 mg·L-1腐植酸作為水溶性有機質代表,考察了其對目標化合物萃取效率的影響。結果表明,腐植酸的存在顯著降低了目標化合物回收率(P<0.05),降低幅度達30%(圖2e)。將萃取質經過C18小柱固相萃取凈化可顯著提升目標化合物回收率(約為103%)。這是由于C18鍵合硅膠柱可有效吸附以去除水樣中的腐植酸成分,從而有效提升目標化合物回收率。本研究發現C18小柱對目標化合物吸附保留作用弱,因此使用其進行固相萃取凈化時無需額外的洗脫過程,可以直接收集萃取質過柱流出液過濾后備測,從而有利于簡化目標化合物萃取工作步驟。通過以上優化實驗獲得了最優水樣中最優的目標化合物萃取條件,即水樣(100 mL)pH值和鹽度分別調整為7.5和0.50%,以二氯甲烷進行3次萃取(5 mL·次-1),萃取質采用C18固相萃取小柱凈化。
2.3 土樣萃取條件優化
對于土壤樣品,通過考察萃取溶劑(正己烷、二氯甲烷和三氯甲烷)、萃取次數以及固相萃取凈化柱以期獲得目標化合物的最優萃取方法。考慮到土壤理化性質尤其是有機質對有機污染物萃取的重要影響,土壤樣品萃取優化實驗同時選用了有機質含量差異較大的兩種土壤即Soil1和Soil 2,它們的有機質含量分別為6.38 g·kg-1和24.1 g·kg-1。優化實驗結果表明,與水樣萃取方法類似,3種萃取劑中二氯甲烷對兩種土壤中目標化合物(1 mg·kg-1)的萃取效率也是最優的,萃取回收率(91.6%-95.8%)高于正己烷和三氯甲烷(30%-75%,圖3a)。就萃取次數而言,當二氯甲烷萃取(5 mL)2次時,兩種土壤中目標化合物的萃取回收率為105%左右,遠高于1次萃取效率(40%-80%),而與3次萃取相當(圖3b)。就萃取凈化柱而言,C18小柱凈化效果最好,目標化合物回收率為94.2%-105.0%,優于WAX柱(88.2%-92.7%)和HLB柱(66.7%-80.1%)。不同凈化柱萃取效率的差異與其固相成分和性能的不同有關,C18柱固定相為十八烷基鍵合硅膠可以有效去除土壤中的基質成分,但對目標化合物保留作用小,因此顯示出高回收率;而HLB柱為親水親油平衡吸附柱,其在去除土壤基質成分的過程中可能還對目標化合物具有一定的吸附作用,從而導致目標化合物回收率較低;而WAX為弱陰離子交換吸附柱,其對目標化合物吸附作用弱,且對土壤基質成分去除作用也相對較弱,所以導致對目標化合物的回收率低于C18柱。由上述優化實驗獲得土樣中目標化合物的最優萃取條件為:取1.0 g土壤,以二氯甲烷提取2次,合并萃取基質濃縮近干并用甲醇定容后過Cl8柱凈化。
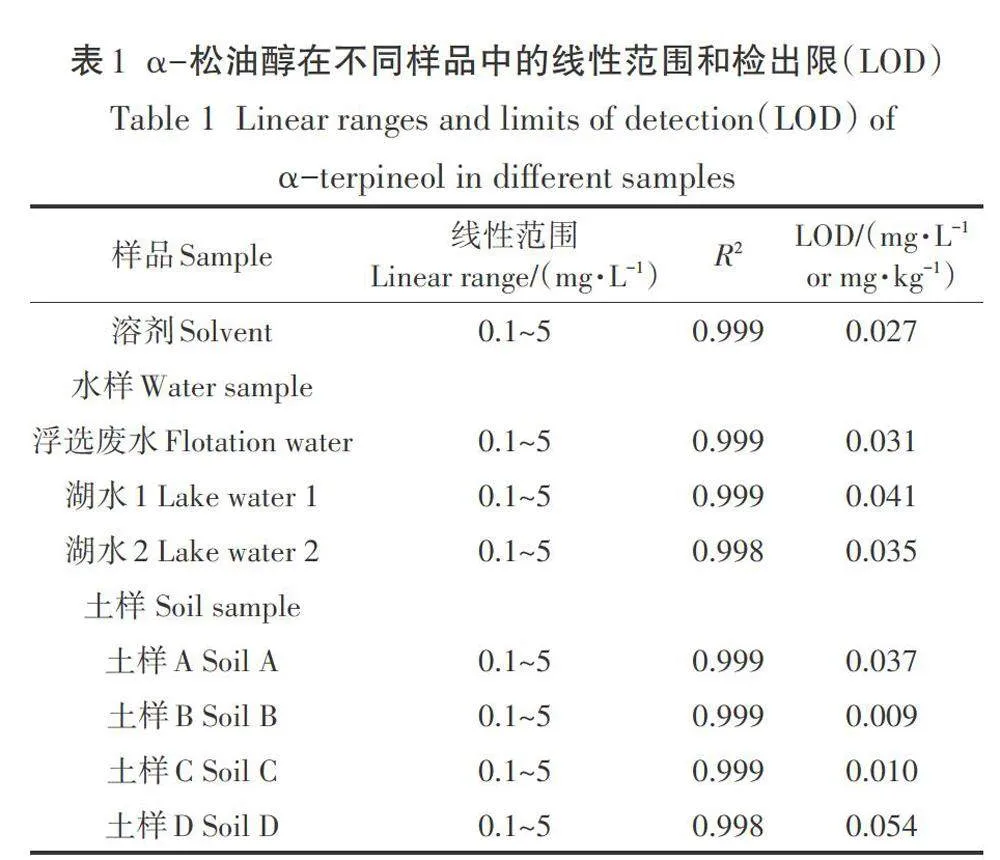
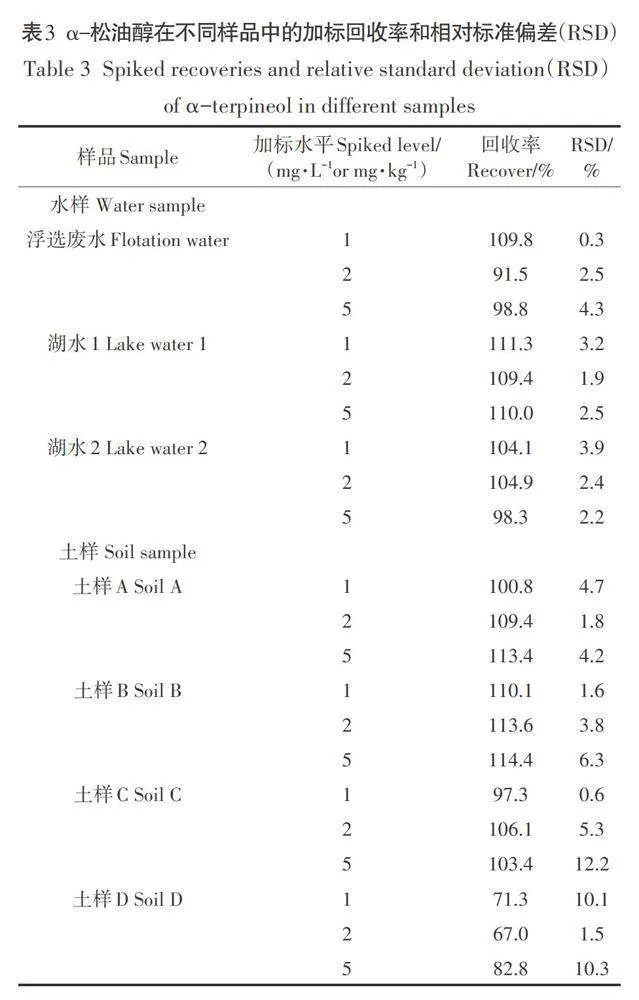
2.4 線性范圍、檢出限、回收率和精密度
在優化條件下,分別測定了不同水樣(湖水、礦區廢棄地浮選廢水)和不同理化性質土樣(4種,分別記作土樣A-土樣D)中目標化合物的線性范圍、檢出限、回收率和精密度。上述樣品均未受到目標化合物污染,其pH值、有機質含量、陽離子交換量(CEC)、黏土含量、砂土含量、粉土含量分別為6.54-8.53、6.38-20.1 g·kg-1、9.1-57.1 cmol· kg-1、1.0% -7.1%、26.4% -84.0%、15.0% - 66.5%。結果表明,不同水樣和土樣基質中目標化合物在實驗濃度范圍(0.1-5 mg·L-1)內均表現出良好的線性關系(R2>0.998,表1),檢出限分別為0.027 mg·L-1(溶劑)、0.031-0.041 mg·L-1(水樣)和0.010-0.054 mg·L-1(土樣),該結果比前人利用GC/MS或HPLC測定水樣、香料植物葉片、工業產品(木樟腦)中目標化合物的檢出限低2-4個數量級(1-610mg·L-1,表2),顯示出本方法的高靈敏度。不同濃度(1、2、5 mg·L-1或1、2、5 mg·kg-1)目標化合物在水樣中的加標回收率為91.5 0-10-11 1.3%,相對標準偏差(RSD)為0.3 %-4.3%;在土樣中的加標回收率為71.3%-114.4%(表3),符合對有機化合物分析國際準則(DG SANC0/12459/2011)的要求,即目標有機化合物回收率在70%-120%之間,RSD<20%。個別土樣回收率略低于70%(67.0%,表3),該結果對于痕量有機污染物分析來說也是可以接受的結果。需要說明的是,盡管前處理過程可有效去除樣品基質成分,但殘留基質成分仍可能影響目標化合物的離子化效率和檢測,導致其檢測信號增加或減弱形成基質增強或基質減弱效應,從而影響目標化合物的準確定量。因此,開展環境樣品尤其是含有復雜基質樣品的測定時,推薦使用樣品基質標線進行定量,以確保目標化合物檢測的準確性。事實上,上述環境樣品中目標化合物加標回收率就是采用基質標線測定獲得的。
3 結論
(1)本研究成功建立了一種基于固相萃取凈化—CC/MS測定水/土樣品中典型浮選藥劑α-松油醇的方法。目標化合物采用EI源-SIM法結合樣品基質標線進行定量分析,定量離子為59,定性離子為93和121。
(2)水樣萃取時,需調節其pH值和鹽度分別為7.5和0.5%,以二氯甲烷進行3次萃取;土壤萃取則以二氯甲烷進行2次萃取。水/土樣品萃取質均采用C18小柱固相萃取凈化。
(3)最優萃取條件下,目標化合物線性范圍為0.1-5 mg·L-1,且具有高靈敏度和滿意的準確度。
(責任編輯:李丹)
基金項目:國家重點研發計劃項目(2020YFC1807600)
