基于PPARs為靶點的抗糖尿病藥物研究進展
2009-04-29 21:32:20何勇彭家志李家明趙永海盛日正馬逢時
亞太傳統醫藥 2009年3期
關鍵詞:糖尿病
何 勇 彭家志 李家明 趙永海 盛日正 馬逢時
(安徽中醫學院 藥學院/安徽省現代中藥重點實驗室,安徽 合肥 230031)お
摘 要:過氧化物酶體增殖因子活化受體(peroxisome proliferator activatived receptors,PPARs)是由配體激活的轉錄因子,屬于核激素受體超家族。PPAR的激活對調節體內的多種代謝過程有重要的作用,被認為是開發治療人類代謝綜合癥藥物的分子靶標,也是目前藥學界研究的熱點。近年來嘗試突破傳統治療藥物的基本結構,研制開發以PPARs為靶點的新型抗糖尿病藥物已成為藥物研究的一大熱點,就PPARs多重激動劑用于治療糖尿病的概況作一綜述。
關鍵詞:過氧化物酶體增殖因子活化受體;糖尿病;代謝綜合癥
中圖分類號:R977.1+5文獻標識碼:A文章編號:1673-2197(2009)03-0129-03
過氧化物酶體增殖因子活化受體(peroxisome proliferator-activated receptor,PPAR)是核受體超家族成員,在控制脂肪的貯藏和分解代謝方面起著重要作用,PPAR存在3種亞型,即PPARα,PPARδ和PPARγ,通過結合特異的DNA序列來調節基因的表達。其中PPARα的激活可以刺激脂肪酸基因的表達和脂質代謝,但對2型糖尿病的治療作用較弱。PPARγ的激活則可以提高胰島素敏感性,減少炎癥的發生,降低游離脂肪酸的脂質濃度及降低血壓,但其對脂代謝紊亂的調節作用較弱,像噻唑烷二酮類藥物有肝臟毒性。最近的研究發現,PPARδ可以控制體重增加,增強身體耐力,提高胰島素敏感性,改善動脈粥樣硬化。因此開發PPARα/PPARγ、PPARα/PPAR δ、PPARγ/PPARδ雙重激動劑和PPARα/PPARγ/PPARδ三重激動劑成為各國學者的研究熱點。
1 PPARα/γ雙重激動劑
與單一的PPARα、PPARγ激動劑相比,PPARα/γ雙重激動劑可以集胰島素增敏作用與降低脂質濃度兩優點于一身,用于治療Ⅱ型糖尿病的高血糖及并發的心血管疾病。因此,研究PPARα/γ雙重激動劑具有更好的開發潛力。Han等人發現α-酰基-β-苯丙酸類衍生物(1)[1],對PPARα、γ的表達EC50分別為19 nM、13 nM,藥效顯示在減少血糖和三酰甘油濃度與有劑量有關;Ye等合成一系列哌啶和脫氫哌啶羧酸衍生物,從構效關系上看1, 3-氧苯基去氫哌啶、1, 4-氧苯基去氫哌啶和1, 3-氧苯基-β, γ-不飽和酸是成為PPARα/γ潛在雙重激動劑重要部位,其中化合物(2)[2]對人類PPARα、γ的表達EC50分別為0.01 μM、0.009 μM;IC50分別為0.861 μM、0.13 μM。另外Parmenon等人以4, 4-二甲基-1, 2, 3, 4-四氫喹啉為先導物合成了對PPARα作用較弱,對PPARγ不完全激動的化合物(3)[3],又在其基礎上對羰基進行改造,并進一步發現化合物(4)[3]對人類PPARα、γ的表達分別為114 nM,7.85nM,對PPARγ的激動程度達95%,具有PPARα/γ雙重激動劑作用;從合成苯丙酸類衍生物(5-8)(4)[4]中發現化合物乙烷基、乙炔基、乙烯基、胺基部分和PPARα和PPARγ基因選擇性粘合時,單鍵化合物能緩和的和聯結點結合在一起,導致5-甲基-2-苯基噁唑的空間立體基團掉入疏水基空腔;使化合物(5)的向酸首基粘結部位移動,這些化合物與這個靶點相互作用,由于殘基的不同和PPARα、PPARγ亞型很好的結合;其中化合物(5)對人類的PPARα、γ的表達IC50分別為185 nM,912 nM;EC50分別為140 nM、623 nM,并對人類PPARγ的激動程度達到94%,其作用機制符合PPARα/γ雙重激動劑的特點,具有成為預防和治療2 型糖尿病藥物的潛力。
2 PPARδ/γ雙重激動劑
由于服用PPARγ激動劑后引起體重增加等不良反應,而PPARδ可以控制體重增加,增強身體耐力,因此在優缺互補的基礎上,Qi等人發現化合物(9)對人類PPARδ/γ的IC50表達分別為5 nM、50 nM;而化合物(10)對人類PPARδ/γ的IC50表達分別為5 nM、39 nM;其中化合物(10)在Zucker diabetic fatty(ZDF)老鼠模型中研究發
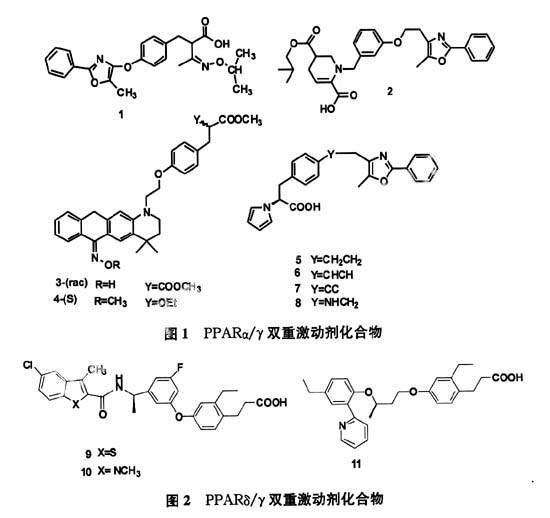
現,與rosiglitazone相比,在相同劑量時,有顯著的降低血糖和減輕rosiglitazone引起體重增加的副作用[5],完全符合雙重激動劑的特點;另外GONZALEZ[6]發現化合物(11)具有PPARδ/γ雙重激動劑的特點,且對人類的PPARδ(IC50=3nM)、PPARγ(IC50=35 nM)基因有高親和性,在細胞轉活試驗中有潛在對抗活性,對ZDF雄鼠,在相同劑量(1 mg/kg)的情況下,化合物(11)在降低血糖和減少體重增加水平上優于rosiglitazone,對ZDF雌鼠,化合物(11)在減少血糖和胰島素方面有顯著作用[6]。
3 PPARα/δ激動劑
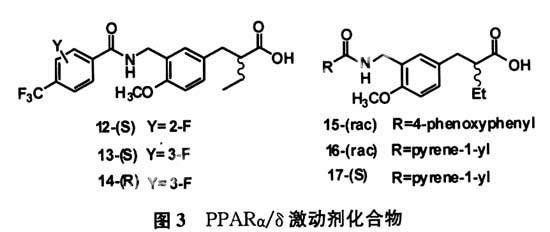
鑒于PPARα和δ激動劑各自作用的特點,發展具有PPARα/δ雙重激動活性新藥已成為熱點的研究領域。該類激動劑同時激活PPARα和PPARδ,理論上應具有各單亞型激動劑的互補優勢,同時避免和減少其存在的一些不良反應。最近KASUGA等人[7-8]發現化合物(12、13、14)對PPARα、PPARδ的表達分別為10 nM,12 nM、12 nM,23 nM、180 nM,700 nM;構效關系表明化合物13(S)構型活性遠遠高于化合物14(R),而F原子在苯環位置的取代活性變化不大。Kasuga等人[9]合成α-取代苯丙酸衍生物像化合物15、16、17,其中對對PPARα、PPARδ的表達分別為8.5 nM,120 nM、24 nM,66 nM、10 nM,40 nM,從構效關系上看化合物(15)對PPARα表達明顯強于PPARδ,而化合物17(S)構型活性高于消旋體,由此看出這類化合物的(S)構型活性高于(R)構型。
4 PPAR的三重激動劑
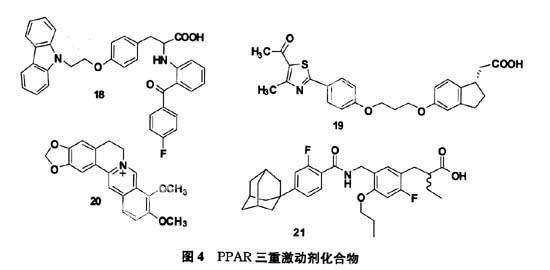
Wang等[10]報道,PPARδ可以緩解PPARγ對脂肪細胞分化的誘導,減少脂肪堆積,因此,PPARα、δ、γ三重激活劑既能提高機體對胰島素的敏感性,又可以通過調節自由脂肪酸、三酰甘油的含量來降低白色脂肪的沉積,可望具有減少心血管并發癥及不誘導肥胖等作用;蘭玉坤等人合成化合物(18)具有PPARα、δ、γ三重激活劑特點[11];另外CANTIN等人發現化合物(19)中的茚滿乙酸基團是一個多用途的酸性首基,來自不同方向的尾基結合與不同的選擇性受體亞型產生PPAR激動作用。最佳的尾部基團使得化合物同時具有PPARδ、γ雙重激動劑和PPARα、δ、γ三重激動劑特點[12]。周吉銀[13]發現中藥毛茛科植物黃連中的小檗堿(20),在給大鼠腹腔注射鏈脲菌素(35 mg/kg) 2 周后,用高糖高脂飼料喂養14 周之后,連續16周分別每天拌食給予小檗堿(150、300 mg/kg)和羅格列酮(4 mg/kg)的治療能增加糖尿病大鼠視網膜的厚度,但視網膜的結構在各組間無差別,小檗堿(150、300 mg/kg)和羅格列酮(4 mg/kg)能明顯降低糖尿病大鼠視網膜中PPAR蛋白表達,小檗堿(150、300 mg/kg)和非諾貝特(100 mg/kg)能顯著增加糖尿病大鼠視網膜中PPARα和PPARδ的表達,小檗堿調控視網膜PPARα、δ、γ蛋白表達可能是其改善糖尿病視網膜病的機制之一,因此可能成為比羅格列酮和非諾貝特更有效地用于治療糖尿病視網膜病的藥物。Kasuga通過研究表明化合物(21)中F原子在適當的位置可以提高目前苯丙酸類PPAR激動劑的活性,其中化合物的(S)構型對PPARα、PPARδ、PPARγ的表達IC50分別為12 nM,25 nM,38 nM遠遠好于(R)構型的200 nM,180 nM,160 nM[14]。
5 展望
針對PPAR各個亞型的化合物結構并非完全獨立,而是具有一定的相關性。隨著對PPAR及相關活性化合物構效關系的深入研究,突破傳統的糖尿病治療藥物基本結構的局限,在活性結構優化的基礎上,結合傳統中藥小分子化合物如小檗堿、白藜蘆醇,我們期望研究開發出更有效、更安全的治療代謝綜合征新藥,有望對代謝綜合征及其并發征的治療帶來深遠的影響。
參考文獻:
[1] HAN H O, KIM S H, KIM K H, et al. Design and synthesis of oxime ethers of α-acyl-β-phenylpropanoicacids as PPAR dual agonists[J]. Bioorg. Med. Chem. Lett,2007, 17(4): 937-941.
[2] YE X Y, LI Y X, FARRELLY D, et al. Design, synthesis, and structure-activity relationships of piperidine and dehydropiperidine carboxylic acids as novel, potent dual PPARα/γagonists[J]. Bioorg. Med. Chem. Lett,2008, 18(12): 3545-3550.
[3] PARMENON C, GUILLARD J, CAIGNARD D H, et al. 4,4-dimethyl-1,2,3,4-tetrahydroquinoline-based PPARα/γ agonists. part I: Synthesis and pharmacological evaluation[J]. Bioorg. Med. Chem. Lett,2008, 18(5): 1617-1622.
[4] CASIMIRO-GARCIA A, BIGGE C F, DAVIS J A, et al. Effects of modifications of the linker in a series of phenylpropanoic acid derivatives: Synthesis, evaluation as PPARα/γdual agonists, and X-ray crystallographic studies [J]. Bioorg. Med. Chem,2008, 16(9): 4883-4907
[5] SHI Q, CANADA E J, XU Y P, et al. Design and synthesis of novel and potent amide linked PPARδ/γ dual agonists[J]. Bioorg. Med. Chem. Lett,2007, 17(24): 6744-6749.
[6] GONZALEZ I C, LAMAR J, IRADIER F, et al. Design and synthesis of a novel class of dual PPARδ/γagonists[J]. Bioorg. Med. Chem. Lett,2007, 17(4): 1052-1055.
[7] KASUGA J I, MAKISHIMA M, HASHIMOTO Y C, et al. Design and synthesis of substituted phenylpropanoic acidderivatives as human peroxisome proliferator-activated receptor α/δ dual agonists [J]. Bioorg. Med. Chem. Lett,2006, 16(3): 554-558.
[8] KASUGA J I, HASHIMOTO Y C, MIYACHI H, et al, Concise and efficient asymmetric synthesis of (S)-2-ethylphenylpropanoic acid derivatives: Dual agonists for human peroxisome proliferator-activated receptorα/δ[J]. Bioorg. Med. Chem. Lett,2006, 16(4): 771-774.
[9] KASUGA J I, YAMASAKI D, ARAYA Y, et al. Design, synthesis, and evaluation of a novel series of a-substituted phenylpropanoic acid derivatives as human peroxisome proliferator-activated receptor (PPAR)α/δdual agonists for the treatment of metabolic syndrome[J]. Bioorg. Med. Chem,2006, 14(24): 8405-8414.
[10] WANG Y X, LEE C H, TIEP S, et al. Peroxisome proliferator-activated receptorδActivates fat metabolism to prevent obesity[J]. Cell, 2003, 113(2): 159-170.
[11] 蘭玉坤, 馬保順, 尹子卉, 等. 新型胰島素增敏劑西格列羧的合成[J]. 中國新藥雜志, 2004, 13(8): 718-720.
[12] CANTIN L D, LIANG S, OGUTU H, et al. Indanylacetic acid derivatives carrying aryl-pyridyl and aryl-pyrimidinyl tail groups-new classes of PPARδ/γand PPAR α/δ/γ agonists[J]. Bioorg. Med. Chem. Lett,2007, 17(4): 1056-1061.
[13] 周吉銀, 周世文. 小檗堿對2型糖尿病視網膜 PPARα/δ/γ表達的影響[J]. 藥學學報,2007, 42(12): 1243-1249.
[14] HOLMES P, MACHER N, GROVE R J, et al. Designing better coumarin-based fluorogenic substrates for PTP1B [J]. Bioorg. Med. Chem. Lett,2008, 18(11): 3382-3385.
(責任編輯:姜付平)
猜你喜歡
中老年保健(2022年5期)2022-08-24 02:35:42
中老年保健(2022年1期)2022-08-17 06:14:56
中老年保健(2021年5期)2021-08-24 07:07:20
中老年保健(2021年9期)2021-08-24 03:51:04
中老年保健(2021年7期)2021-08-22 07:42:16
中老年保健(2021年3期)2021-08-22 06:49:56
中老年保健(2021年11期)2021-08-22 03:15:16
中國生殖健康(2020年2期)2021-01-18 02:51:44
中國生殖健康(2018年2期)2018-11-06 07:11:04
基層中醫藥(2018年2期)2018-05-31 08:45:04
