熒光法測定鈾溶液中的微量釷
2010-01-26 08:15:29錢紅娟張麗華趙立飛吳繼宗劉煥良
核化學與放射化學 2010年4期
錢紅娟,張麗華,蘇 濤,趙立飛,吳繼宗,劉煥良
中國原子能科學研究院 放射化學研究所,北京 102413
釷是核產品中需要嚴格控制的雜質元素之一,需要準確測定。建立一個靈敏度高、選擇性好的微量釷分析方法很有意義。核領域的分析樣品體系都很復雜,同時具有強放射性和高毒性,樣品中大量存在的鈾、钚、稀土、鋯等元素嚴重干擾釷的測定。因此,樣品分析前,需對其進行預處理。
釷在水溶液中以四價形式存在,在分離和測定時易受钚、鋯等四價元素干擾。釷的分離方法有萃取分離法、離子交換法、萃取色層法等。萃取色層分離法是繼萃取分離和離子交換分離法之后的一種重要的分離手段,其特點是把萃取劑的高選擇性和樹脂填充床的高效性有機地結合,分離速度快,其固定相有更好的耐輻照穩定性等,更適用于熱室中分離強放射性核素。
釷的測定方法有原子發射光譜法[1]、質譜法、同位素稀釋質譜法、光度法[2-3]、熒光光度法[4-5]等。原子發射光譜法是核材料中雜質分析最常用的方法,但釷的光譜譜線多而復雜,分析方法難以掌握;質譜法靈敏度高,但基體影響大,且在測量時需要將元素離子化,儀器必須嚴格密封;同位素稀釋質譜法分析速度慢、分析成本高;分光光度法是我國過去采用的方法,靈敏度僅為mg/L級水平,樣品取樣量大,選擇性不夠理想,尤其是Zr、Hf的干擾問題始終未能解決。熒光光度法測定釷靈敏度很高,可達到μg/L級水平,測定產品中痕量釷有很大的優越性,完全能夠滿足核材料中雜質分析對于靈敏度的要求。
桑色素熒光法是測定釷的傳統方法,但桑色素易被氧化,熒光強度不穩定,干擾元素較多,在分析中應用受到限制。釷-桑色素(Morin)-三辛基氧膦(TOPO)形成的多元絡合物,具有發強烈熒光、發色速度快、絡合物穩定、靈敏度高等優點,更適合于釷的熒光測定。文獻[5]曾用該體系測定礦石和礦渣中的微量釷,利用CL-5209萃淋樹脂分離去除基體干擾,采用HCl洗脫液,但HCl具有強腐蝕性,在后處理分析中應盡量避免;采用緩沖體系進行熒光測定,絡合物的熒光強度明顯低于HNO3體系。本工作擬在HNO3體系中采用Th-桑色素-TOPO三元絡合物熒光法測定Th。
1 試驗部分
1.1 儀器與試劑
850熒光分光光度計,日本日立公司;F96熒光分光光度計,上海棱光儀器公司;ICP-AES,美國熱電公司。
CL-TBP萃淋樹脂,TBP質量分數60%,粒度0.125~0.163 mm;標準釷溶液,核工業化工冶金研究院;桑色素、三辛基氧膦、十二烷基磺酸鈉溶液(SLS)、聚乙二醇辛基苯基醚(Tritonx_100),分析純,均為北京化學試劑公司;H2SO4、HNO3、HCl均為優級純試劑。
1.2 實驗步驟
1.2.1分離柱的制備 CL-TBP萃淋樹脂浸泡24 h以上,采用濕法裝柱。用20 mL 1.0 mol/L HNO3洗滌柱子,再用水洗至中性;用20 mLw=5% Na2CO3洗滌,水洗至中性,待用。
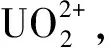
1.2.3樣品測量 準確移取一定量分離后的樣品于10 mL容量瓶,加入1.5 mL 2.5 mol/L HNO3、1.5 mLw=0.01% TOPO、2.0 mLw=0.02%桑色素、1.5 mLw=0.25% SLS,用水定容,混合均勻。放置15 min后,在激發波長420 nm、發射波長505 nm處測量絡合物的熒光強度。
2 結果與討論
2.1 分離方法

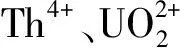
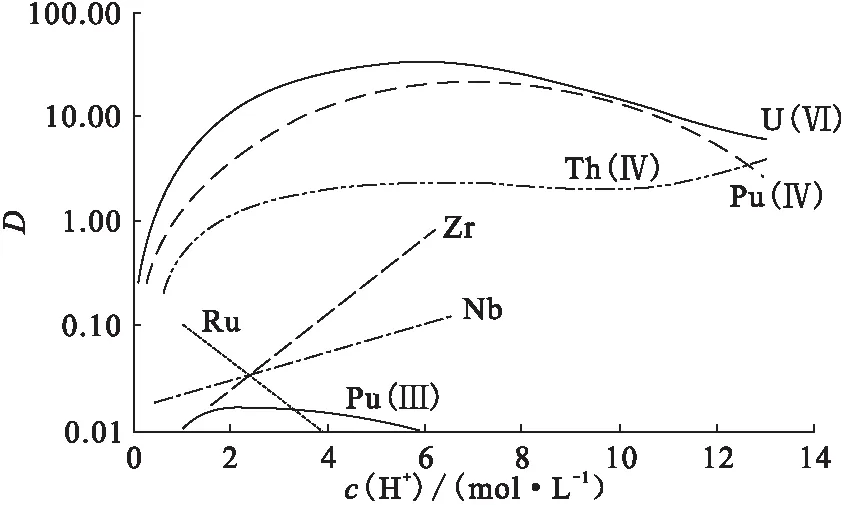
圖1 鈾、钚、釷等在硝酸溶液和CL-TBP上的分配比[1]Fig.1 The partition ratio between in nitric acid solution and CL-TBP[1]
2.1.2萃取色層柱的制備 試驗過程中,分離柱的流速需要控制。分離柱的流速較慢,會使實驗周期過長;若流速過快,溶液與樹脂床層接觸時間短,不利于萃淋樹脂對金屬離子的吸附,影響分離效果。本試驗分離柱的流速控制為1.0~1.5 mL/min。考察了粒徑為0.250~0.420、0.194~0.250、0.125~0.163 mm萃淋樹脂的流速,流速分別為3.5、2.0、1.5 mL/min。本試驗選用粒徑為0.125~0.163 mm的萃淋樹脂。經試驗確定的萃取色層柱示于圖2。
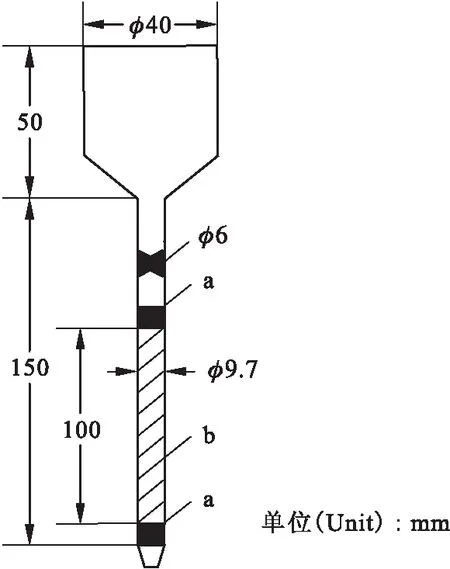
圖2 萃取色層柱(φ6 mm×100 mm,玻璃材料)Fig.2 Extraction column(φ6 mm×100 mm,glasses)a——聚四氟乙烯絲(Polyflon funicle);b—— CL-TBP萃淋樹脂(CL-TBP resin)
2.1.3Th(Ⅳ)吸附及洗脫條件的選擇
(1) 酸度對分配比的影響

(2) 吸附條件的選擇
實現Th4+和Zr4+、Fe3+有效分離的關鍵,就是選擇合適的吸附條件。從圖1可見,當c(HNO3)>4.0 mol/L(特別是c(HNO3)達到6.0 mol/L)時,Zr4+在CL-TBP萃淋樹脂具有一定的吸附能力。為了使Zr4+、Fe3+等離子,特別是Zr4+不被CL-TBP萃淋樹脂所吸附,同時又確保Th4+在樹脂上的吸附率,吸附酸度不能高于3.0 mol/L HNO3。
分別考察Th4+在2.0 mol/L和3.0 mol/L HNO3條件下在樹脂上的吸附性能。實驗結果表明,在2.0 mol/L和3.0 mol/L HNO3介質條件下,Th4+均能完全吸附于CL-TBP樹脂上。實驗中選擇2.0 mol/L HNO3吸附Th4+。
(3) 洗脫條件的選擇
(4) 淋洗曲線
取50 μg Th4+通過分離柱,每次用1 mL 0.4 mol/L HNO3洗脫,收集洗脫液,繪制淋洗曲線(圖3)。對于50 μg Th4+,8 mL 0.4 mol/L HNO3可完全洗脫,實驗中采用10 mL 0.4 mol/L HNO3洗脫Th4+。

圖3 淋洗曲線Fig.3 Leaching curve
2.1.4共存離子的吸附及淋洗
(1) 鈾、釷的分離
(2) 其它離子的吸附及淋洗
不同離子依次上柱,分別用10 mL 2.0 mol/L HNO3淋洗分離柱,測定淋洗液中不同離子的濃度,結果列于表1。
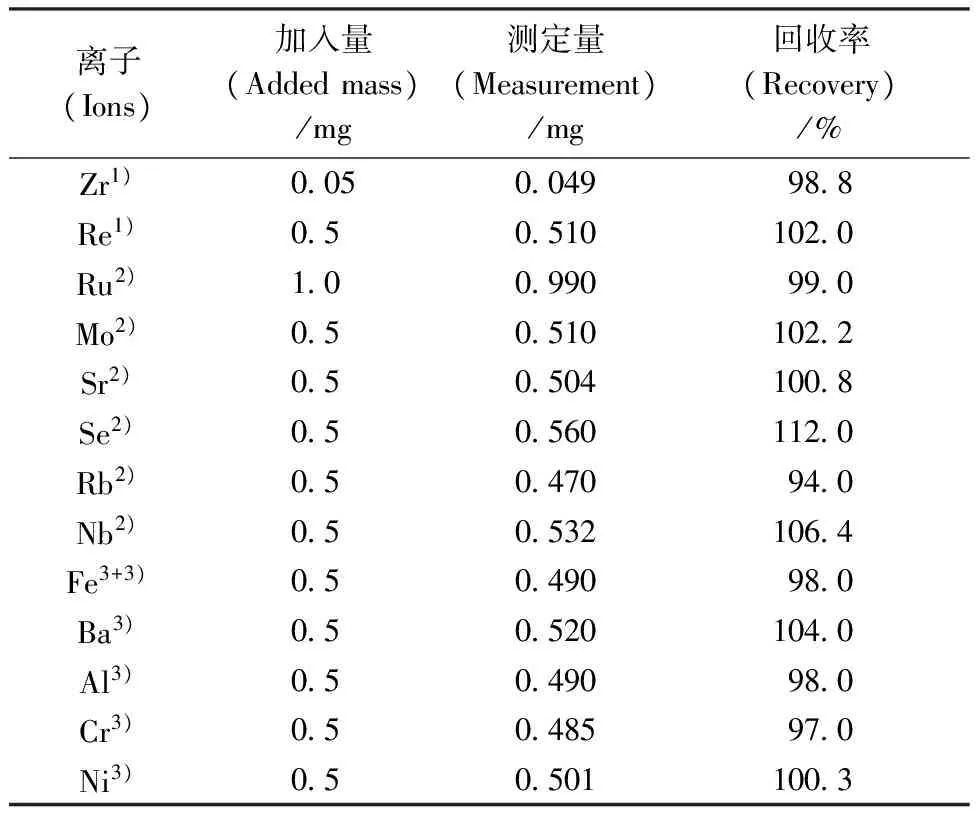
表1 共存離子的吸附和淋洗回收率Table 1 Absorption and leaching of coexisting-ions
注:1) 分光光度法(Spectrophotometry)
2) X-射線熒光法(X-ray fluorescence method)
3) ICP-AES
表1結果表明,在2.0 mol/L HNO3介質中,Zr、Re、Sr、Al、Fe、Se、Ru、Nb、Mo、Rb等離子在CL-TBP萃淋樹脂上均無吸附,且用10 mL 2.0 mol/L HNO3都可淋洗完全。
2.1.5考驗Th、U、Zr的分離效果 配制Th、U、Zr合成樣品,其質量濃度分別為10、2 000、50 mg/L。取1 mL合成樣品上柱,10 mL 2.0 mol/L HNO3淋洗;10 mL 0.4 mol/L HNO3洗脫,洗脫液收集于容量瓶中,測量洗脫液成分含量,結果列于表2。從表2結果可以看出,Th的回收率在95.6%~102%之間;Th與Zr、U可以完全分離。
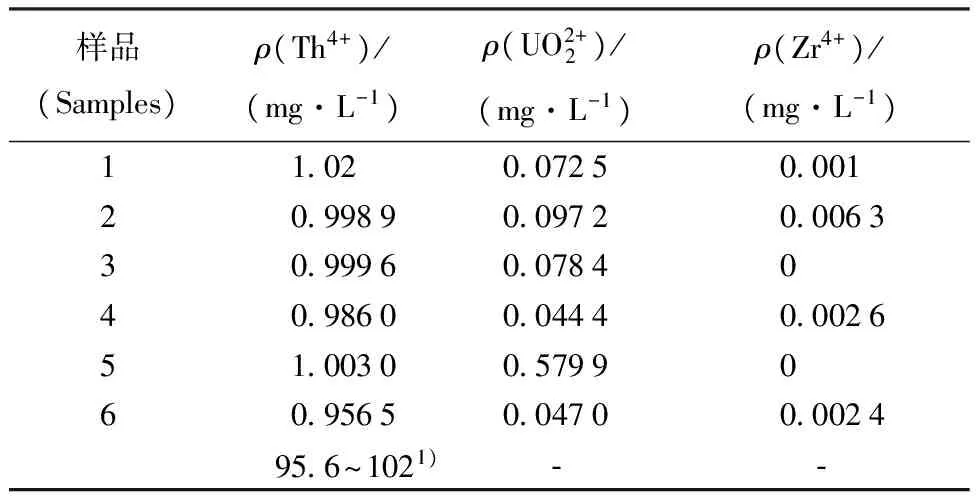
表2 洗脫液的ICP-AES分析結果Table 2 Determination results of synthetic sample by ICP-AES
注(Note):1) Th的回收率(Recovery of Th)
2.2 分析方法
2.2.1激發波長和發射波長 釷-桑色素-三辛基氧膦形成的多元絡合物,具有熒光強、發色速度快、絡合物穩定等優點。實驗選用釷-桑色素-三辛基氧膦熒光體系進行微量釷的測定。絡合物的熒光強度不僅與激發光強度有關,而且與熒光發射時的量子產率有關。在激發光譜的波峰處,能獲得最強的激發光;在發射光譜的波峰處,能獲得最大量子產率。圖4為絡合物的激發光譜和發射光譜,選擇最佳的激發波長為420 nm,發射波長為504 nm。
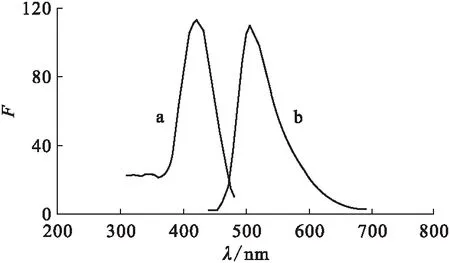
圖4 發射光譜(a)和激發光譜(b)Fig.4 Excitation(a) and emission(b)
2.2.2酸度影響
(1) 酸體系的選擇
比較了H2SO4、HCl、HNO3、H3PO4體系對絡合物熒光強度的影響,試驗表明,釷-桑色素-TOPO三元絡合物在H2SO4、HCl、HNO3、H3PO4介質的熒光強度(F)無顯著差異。在HNO3介質中,試劑空白最小,有利于提高方法的靈敏度,因此選擇HNO3介質。
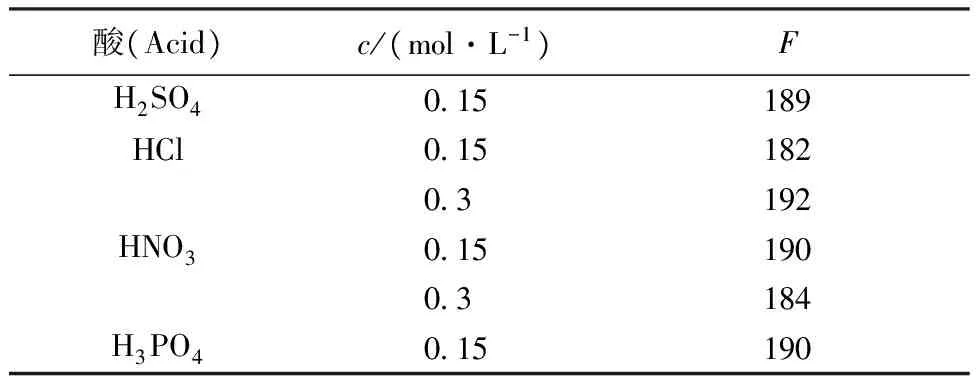
表3 酸對絡合物熒光強度的影響Table 3 Effect of acid on fluorescent intensity
注(Note):m(Th4+)= 50 ng
(2) 酸度影響
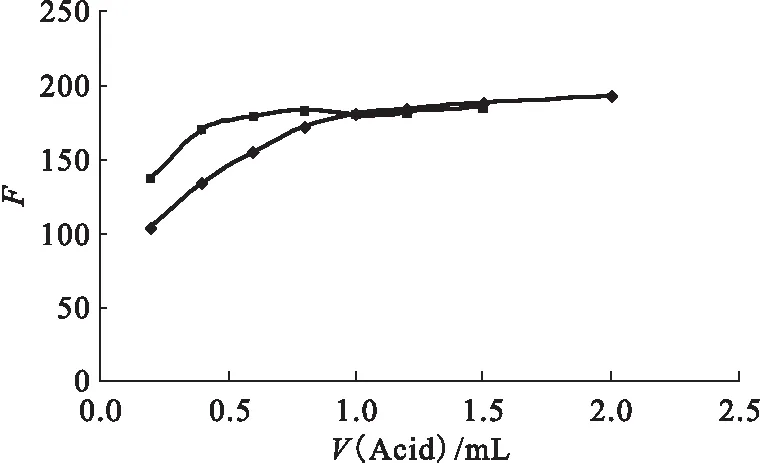
圖5 酸度對絡合物熒光強度的影響Fig.5 Effect of acidity on fluorescent intensity■——2.5 mol/L H2SO4;◆——2.5 mol/L HNO3
影響絡合反應平衡的因素很多,溶液酸度是最重要因素之一。溶液酸度的影響主要表現為H+與金屬離子的競爭反應。圖5為溶液酸度對絡合物熒光強度的影響曲線。從熒光強度-酸度曲線來看,熒光強度隨著溶液酸度的增加而增大;當溶液酸度達到0.25 mol/L HNO3,絡合物熒光強度趨于穩定。本工作選擇體系酸度為0.3 mol/L。
2.2.3SLS溶液用量的選擇 釷-桑色素-三辛基氧膦多元絡合物為非水溶性物質,利用表面活性劑(SLS)膠束增容作用,使絡合反應直接在水相中進行。改變SLS的加入量,觀察SLS用量對熒光強度的影響,結果示于圖6。從圖6可見,0.25% SLS加入量在1.2~2.5 mL范圍內,絡合物的熒光強度趨于穩定。試驗中0.25% SLS的加入量為2.0 mL。
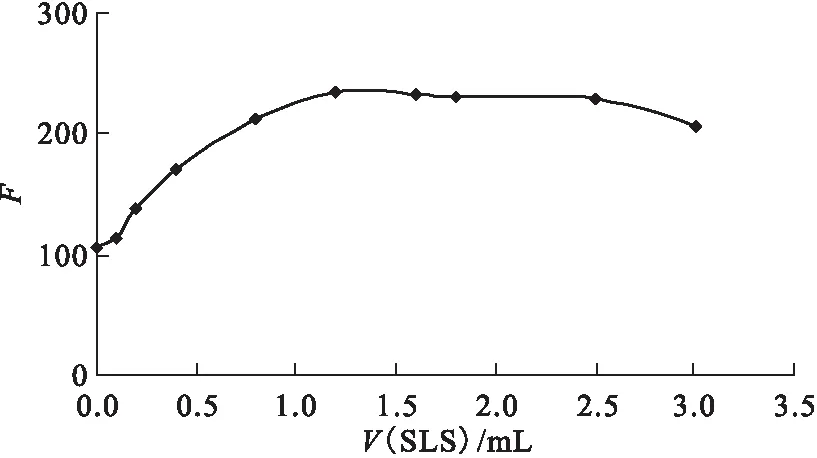
圖6 SLS溶液用量對絡合物熒光強度的影響Fig.6 Effect of SLS on fluorescent intensity
2.2.4TOPO用量的選擇 改變TOPO的加入量,觀察TOPO用量對熒光強度的影響,結果示于圖7。從圖7可見,3×10-4mol/L TOPO加入體積達到0.8 mL時,絡合物的熒光強度趨于穩定。試驗中選擇3×10-4mol/L TOPO的加入量為1.5 mL。
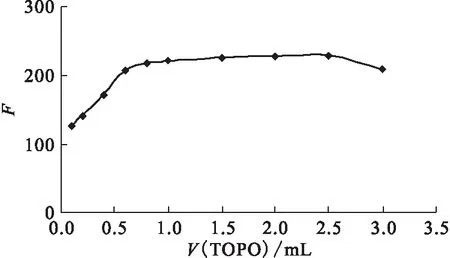
圖7 TOPO用量對絡合物熒光強度的影響Fig.7 Effect of TOPO on fluorescent intensity

圖8 桑色素用量的選擇Fig.8 Effect of morin on fluorescent intensity
2.2.5桑色素用量的選擇 桑色素用量過大,會增加試劑空白值。用量不足,會影響多元絡合物的形成。桑色素用量對絡合物吸光度的影響曲線示于圖8。由圖8可見,0.02%桑色素的適宜用量為2.0 mL。
2.2.6反應時間的影響 反應平衡時間受溫度影響,在室溫~15 ℃時,繪制反應時間對吸光度的影響曲線示于圖9。由圖9可見,絡合反應可在10 min內達到平衡,而且絡合物的熒光強度在10 h內保持穩定。本實驗選擇反應時間為15 min。
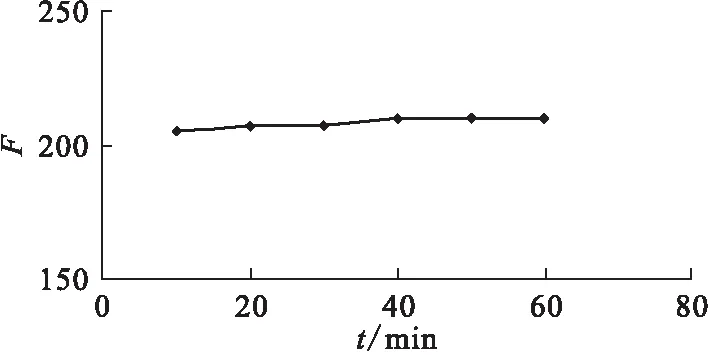
圖9 時間影響曲線Fig.9 Effect of reaction time on fluorescent intensity
2.2.8工作曲線 分別移取不同量的釷標準溶液,在選定的工作條件下繪制工作曲線(圖10)。實驗結果表明,在0~500 μg/L 范圍內,熒光強度與Th含量呈線性關系,檢出限為0.1 μg/L。1.0 μg/L和50.0 μg/L釷標準溶液的測量精密度分別為2.5%、0.6%。
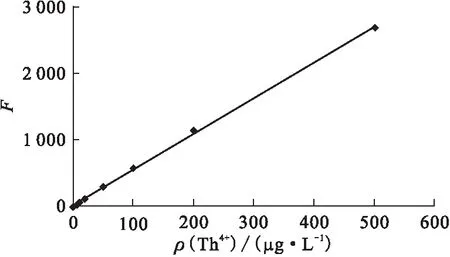
圖10 釷的工作曲線Fig.10 Calibration curve for determination of Th4+
2.3 樣品分析
按表4配制模擬樣品,按所建立的方法分析處理樣品。分析結果與ICP-AES測定結果進行比對,結果列于表5。
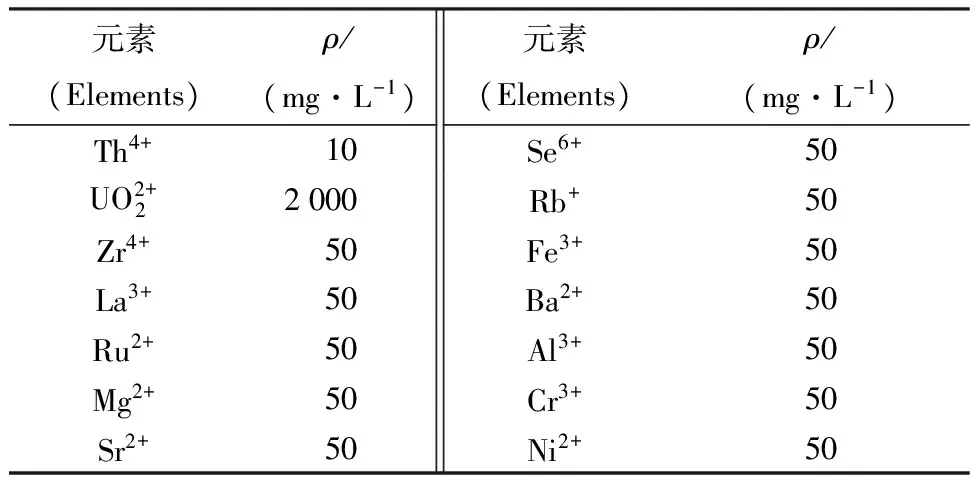
表4 模擬樣品的成分Table 4 Composition of simulate samples
注(Note):c(HNO3)=2.0 mol/L
從表5可以看出,兩種分析方法的分析結果在誤差范圍內保持一致,說明本方法準確、可靠。
3 結 論
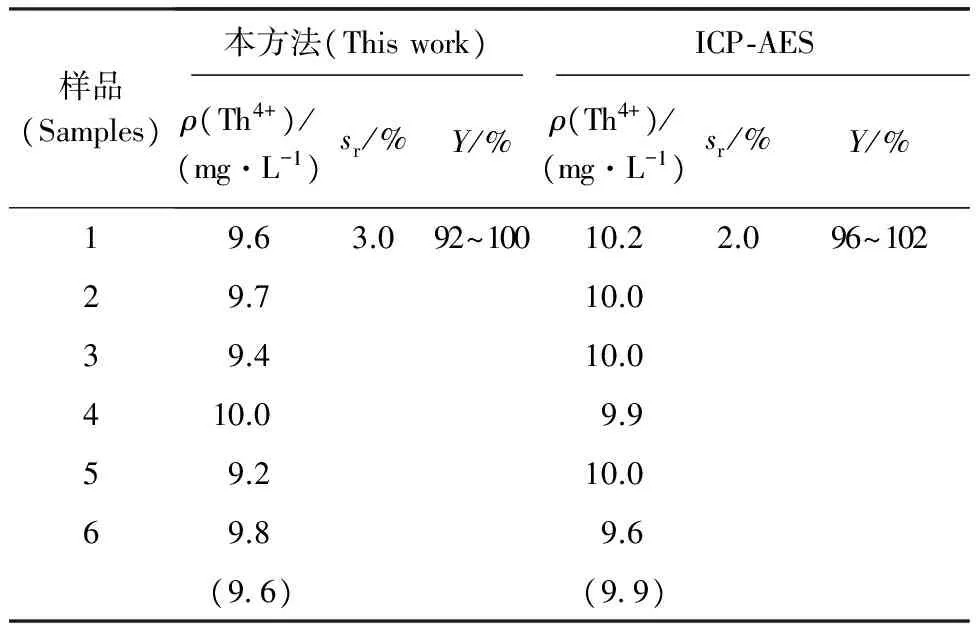
表5 模擬樣品測定結果Table 5 Analytical results of simulate samples
注(Note):括號中數值為平均值(The data in parentheses are the average)
(2) 研究了釷-桑色素-三辛基氧膦熒光體系,確定了熒光測定釷的最佳條件:熒光體系酸度為0.375 mol/L,絡合反應很快達到平衡,激發波長和發射波長分別為420 nm和504 nm,線性范圍0~500 μg/L,檢測限為0.1 μg/L,對于1.0 μg/L和50.0 μg/L樣品,測量精密度分別為2.5%和0.6%。
(3) 本文建立的分析方法,靈敏度高、選擇性好,適用于鈾中微量釷的分析。對模擬樣品的分析結果相對標準偏差為3%,回收率為92%~100%。
[1] 楊正華,戢 素.ICP-AES測定鈾和鈾化合物中微量釷及稀土分段元素[J].光譜學與光譜分析,1989,9(6):74-77.
[2] 王木林,張曉軍,劉 宏.DCS-偶氮胂Ⅲ分光光度法測定釷的研究與應用:稀土精礦中微量釷的測定[J].內蒙古大學學報(自然科學版),1997,28(1):138-140.
[3] Shvoeva O P, Dedkova V P, Savvin S B. Sorption-Spectrometric Determination of Thorium With Arsenazo Ⅲ on a Solid Phase[J]. Anal Chem, 1998, 53(5): 412-415.
[4] 葉開明,岳 峰.熒光光度法測定礦石和礦渣中微量釷[J].鈾礦冶,1996,15(3):183.
[5] 鄧 剛.熒光光度法測定巖石中的微量釷[J].分析試驗室,1988,7(1):25-27.
[6] 宋金如,龔治湘,祝旭初.CL-5209萃淋樹脂吸附釷的性能和機理研究[J].華東地質學院學報,2000,23(4):271-276.
