TiO2對 Ni-Mo/MCM-41催化劑加氫脫硫性能的影響
2010-01-29 02:10:28吳春雷王安杰
石油學報(石油加工) 2010年2期
吳春雷,李 翔,2,王安杰,2
(1.大連理工大學精細化工國家重點實驗室,遼寧大連116012;2.遼寧省高校石油化工技術與裝備重點實驗室,遼寧大連116012)
在傳統的以γ-Al2O3作載體的雙金屬硫化物加氫脫硫(HDS)催化劑中,TiO2具有一定的助劑作用。它的引入一方面能夠提高催化劑酸性[1],提高活性組分分散度[2]以及減弱活性組分與γ-Al2O3之間的相互作用,從而提高活性組分的可還原性和硫化能力[3];另一方面還起到了電子型助劑作用[4]。MCM-41介孔分子篩因其具有比表面積大、孔徑均一、可調等特點,在大分子參與的非均相催化反應中展現出良好的應用前景[5]。筆者[6-7]前期研究結果表明,全硅MCM-41(Si-MCM-41)的硅質骨架結構使得它與活性組分間的相互作用適中,而其較大的比表面積和規整的一維介孔孔道則有利于活性組分分散和大分子含硫化合物的擴散,因而是一種優良的加氫脫硫催化劑載體。其擔載制備的Co-Mo和Ni-Mo催化劑對于二苯并噻吩(DBT)的 HDS反應表現出了很高的催化活性。為了進一步提高Si-MCM-41擔載的Ni-Mo催化劑的 HDS反應性能,以及探討 TiO2的助劑作用,筆者采用分步浸漬的方法在 Ni-Mo/MCM-41催化劑中引入少量的TiO2,以DBT作為模型化合物,考察了 TiO2及其引入順序對Ni-Mo/MCM-41催化劑加氫脫硫反應催化性能的影響。
1 實驗部分
1.1 原料
鉬酸銨((NH4)6Mo7O24·4H2O)、無水乙醇、鈦酸四丁酯(C16H36O4Ti)和硝酸鎳(Ni(NO3)2·6H2O)均為分析純。五水偏硅酸鈉(Na2SiO3·5H2O)為工業級。十六烷基三甲基溴化銨(CTAB)為南京旋光科技有限公司產品,工業級。DBT由聯苯和硫合成[8]。十氫萘購自上海試劑分裝廠,純度>99%。
1.2 MCM-41的合成和催化劑的制備
按文獻[9]方法制備Si-MCM-41。在Na2SiO3·5H2O中加入 H2SO4,調節p H值至11。按一定比例加入模板劑CTAB,在不銹鋼壓力釜中于120℃下晶化48 h。固體經過濾、洗滌、烘干后,于600℃和 N2保護下焙燒12 h,然后在空氣中540℃焙燒6 h,得到Si-MCM-41。
采用共浸漬法制備 Si-MCM-41擔載的 Ni-Mo催化劑。將計量的Si-MCM-41加入到鉬酸銨和硝酸鎳的水溶液中,在室溫下浸漬8 h,所得固體產物在120℃下烘干12 h,在500℃空氣中焙燒3 h,得到Ni-Mo/MCM-41催化劑。
采用分步浸漬的方法制備2種 TiO2改性的Ni-Mo/MCM-41催化劑。
為了得到 TiO2先于活性組分引入的催化劑,首先將Si-MCM-41加入到計量的鈦酸四丁酯的無水乙醇溶液中,在室溫下浸漬8 h,所得固體產物在120℃下烘干 12 h,500℃空氣中焙燒 3 h,得到TiO2-MCM-41載體。然后將計量的 TiO2-MCM-41加入到鉬酸銨和硝酸鎳的水溶液中,在室溫下浸漬8 h,120℃下烘干12 h,500℃空氣中焙燒3 h,得到的催化劑記為Ni-Mo/TiO2-MCM-41。
為了得到在活性組分之后引入 TiO2的催化劑,將制備的Ni-Mo/MCM-41催化劑的前驅體加入到計量的鈦酸四丁酯的無水乙醇溶液中,室溫浸漬8 h后,經120℃烘干12 h,然后在500℃下焙燒3 h,得到的催化劑記為 TiO2-Ni-Mo/MCM-41。
在上述3種催化劑中,擔載MoO3的質量分數均為20%,助劑Ni與Mo摩爾比為0.75,TiO2的質量分數為2%。
1.3 HDS反應
在內徑為8 mm的不銹鋼固定床中壓反應器中進行 HDS反應。催化劑經壓片,破碎至0.5~0.8 mm裝填,用量0.1 g。HDS反應前,用含 H2S體積分數為10%的 H2S-H2混合氣對氧化態催化劑進行硫化,硫化溫度400℃,時間3 h。然后在壓力4.0 MPa、氫/油體積比750、反應溫度280℃的條件下進行 HDS反應。原料為質量分數0.8%的DBT/十氫萘溶液,反應停留時間(Reaction time,定義為催化劑質量(g)/反應物摩爾流量(mol/min))在12.5~50.3(g·min)/mol間變化。采用 Agilent 6890N型氣相色譜儀,配有固定相為(5%)二苯基-(95%)二甲基聚硅氧烷的市售 HP-5型毛細管色譜柱,測定原料和反應產物的組成。
圖1示出了DBT的 HDS反應網絡[10]。DBT的 HDS反應主要通過直接脫硫(DDS)和預加氫脫硫(HYD)兩條并行的反應路徑進行。DDS反應路徑的產物為聯苯(BP),在有機硫化物如DBT存在的情況下,BP很難進一步加氫生成苯基環己烷(CHB)[11]。DBT的 HYD反應路徑可以看作是1個串連反應,四氫硫芴 (TH-DBT)和六氫硫芴(HH-DBT)是 HYD反應路徑的主要含硫中間體,脫硫后生成 CHB,再進一步加氫生成聯環己烷(BCH)。在酸性較強的催化劑上,CHB和BCH還會發生加氫裂化反應,產物為苯(B)和環己烷(C)。在本研究所述實驗條件下,沒有檢測到 TH-DBT、HH-DBT等含硫中間體,因此可以用DBT的轉化率作為催化劑 HDS反應活性的指標。
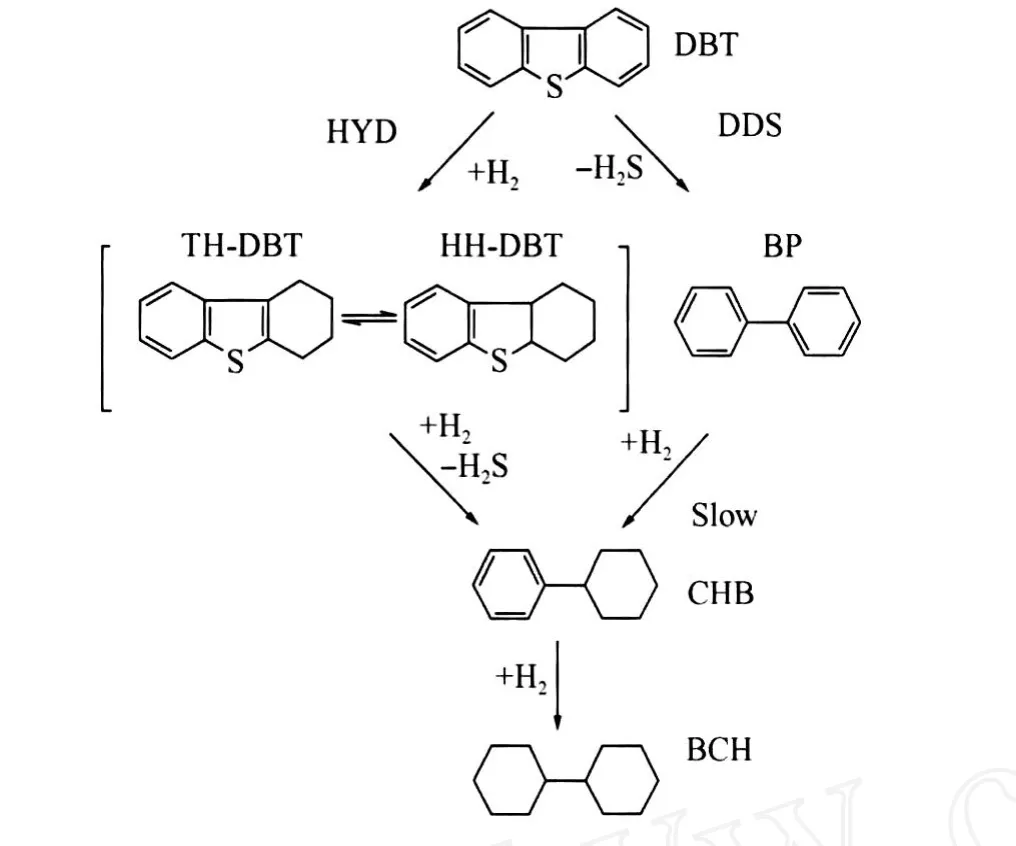
圖1 二苯并噻吩(DBT)的 HDS反應網絡Fig.1 HDS reaction network of dibenzothiophene(DBT)
1.4 催化劑表征
采用日本分光公司Jasco V-550型紫外/可見分光光度計測定催化劑前驅體的紫外/可見漫反射光譜(UV-Vis),掃描范圍190~800 nm。在 Chembet-3000分析儀上進行 TPR試驗。測試前,樣品首先在 He氣氛下200℃處理2 h。還原氣為10%H2+ 90%Ar混合氣(體積分數),升溫速率10℃/min。在Chembet-3000分析儀上用 NH3-TPD測定催化劑前驅體的表面酸強度分布。將0.2 g樣品放在U型石英玻璃管中,500℃下 He氣吹掃0.5 h,然后降溫至150℃并吸附 NH3至飽和,待基線平穩后以18℃/min速率升溫至550℃,He流速20 mL/min。
2 結果與討論
2.1 Ni-Mo/TiO2-MCM-41和 TiO2-Ni-Mo/MCM-41催化劑的表征結果
圖2示出了 Ni-Mo/MCM-41、Ni-Mo/TiO2-MCM-41和 TiO2-Ni-Mo/MCM-41氧化物前驅體的UV-Vis譜圖。在Ni-Mo/MCM-41氧化態前驅體的UV-Vis譜中,250和300 nm處的吸收峰分別對應四面體配位的 MoO2-4和八面體配位的 Mo6+物種[12-13],400~460 nm處的吸收峰對應八面體配位的 Ni2+物種[14]。Ni-Mo/TiO2-MCM-41和 Ni-Mo/ MCM-41的 UV-Vis曲線相差不大,說明 TiO2先于活性組分引入對Ni和Mo物種的配位狀態影響不大。但TiO2-Ni-Mo/MCM-41與Ni-Mo/MCM-41的 UV-Vis相比,TiO2-Ni-Mo/MCM-41在400~460 nm處的紫外吸收峰強度降低,同時300 nm處的八面體配位的Mo6+物種的紫外吸收峰有所增強,說明 TiO2-Ni-Mo/MCM-41前驅體中八面體配位的Ni2+物種含量有所減少,而八面體配位的 Mo6+物種含量有所增加。
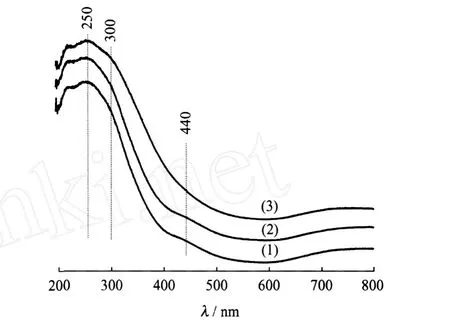
圖2 Ni-Mo/MCM-41、Ni-Mo/TiO2-MCM-41和TiO2-Ni-Mo/MCM-41氧化物前驅體的 UV-Vis譜圖Fig.2 UV-Vis patterns of Ni-Mo/MCM-41,Ni-Mo/ TiO2-MCM-41 and TiO2-Ni-Mo/MCM-41 oxidic precursors(1)Ni-Mo/MCM-41;(2)Ni-Mo/TiO2-MCM-41; (3)TiO2-Ni-Mo/MCM-41
圖3為Ni-Mo/MCM-41、Ni-Mo/TiO2-MCM-41和 TiO2-Ni-Mo/MCM-41氧化態前驅體的 TPR譜圖。Ni-Mo/MCM-41前驅體的 TPR譜圖中主要有2個氫耗峰:403℃的低溫氫耗峰歸屬為八面體配位的 Mo6+物種的部分還原(Mo6+→Mo4+)[15-16], 500℃左右的肩峰可能與α-NiMoO4相中 Ni物種的還原有關[17]。與 Ni-Mo/MCM-41相比,Ni-Mo/ TiO2-MCM-41的 TPR譜圖中低溫氫耗峰的還原溫度和肩峰強度都有所降低,說明將 TiO2先于活性組分引入到MCM-41表面在一定程度上促進了前驅體的還原。這可能是因為 TiO2的引入增加了活性組分與載體之間的相互作用力,提高了活性組分的分散度[18-19]。而 TiO2-Ni-Mo/MCM-41的低溫氫耗峰及其肩峰的還原溫度分別比Ni-Mo/MCM-41相應氫耗峰的溫度升高了5℃和23℃,在一定程度上抑制了催化劑中活性物種的還原。但總的說來,這3種催化劑的 UV-Vis和 TPR譜圖差別并不明顯,說明 TiO2的引入對Ni-Mo/MCM-41催化劑活性物種的配位狀態和還原性能影響不顯著。

圖3 Ni-Mo/MCM-41、Ni-Mo/TiO2-MCM-41和TiO2-Ni-Mo/MCM-41氧化物前驅體TPR譜圖Fig.3 TPR profiles of Ni-Mo/MCM-41,Ni-Mo/TiO2-MCM-41 and TiO2-Ni-Mo/MCM-41 oxidic precursors (1)Ni-Mo/MCM-41;(2)Ni-Mo/TiO2-MCM-41; (3)TiO2-Ni-Mo/MCM-41
圖4為Ni-Mo/MCM-41、Ni-Mo/TiO2-MCM-41和TiO2-Ni-Mo/MCM-41氧化態前驅體的NH3-TPD譜圖。根據 NH3的脫附溫度,可將酸中心大致分為弱酸中心(150~250℃)、中強酸中心(250~350℃)和強酸中心(350~450℃)3類[20]。3種催化劑前驅體在230℃左右可以觀察到明顯的NH3脫附峰,可見3種催化劑前驅體表面主要為弱酸中心。此外,除 Ni-Mo/TiO2-MCM-41的 NH3脫附溫度略有升高外,3者的 NH3-TPD譜圖差別不大,說明引入 TiO2對Ni-Mo/MCM-41催化劑前驅體表面酸性影響不大。
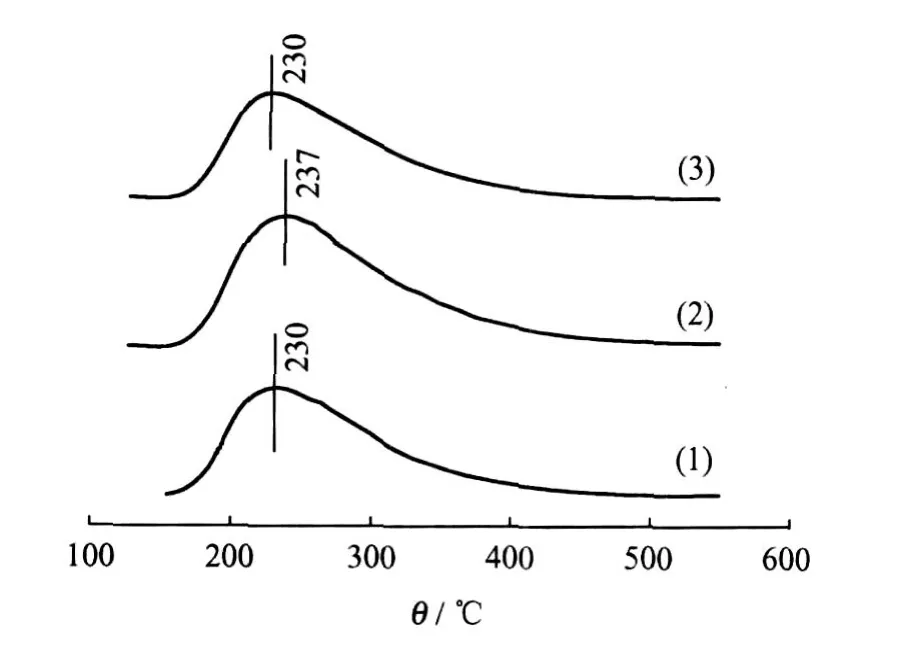
圖4 Ni-Mo/MCM-41、Ni-Mo/TiO2-MCM-41和TiO2-Ni-Mo/MCM-41氧化物前驅體 NH3-TPD譜圖Fig.4 NH3-TPD profiles of Ni-Mo/MCM-41,Ni-Mo/ TiO2-MCM-41 and TiO2-Ni-Mo/MCM-41 oxidic precursors(1)Ni-Mo/MCM-41;(2)Ni-Mo/TiO2-MCM-41; (3)TiO2-Ni-Mo/MCM-41
2.2 Ni-Mo/TiO2-MCM-41和 TiO2-Ni-Mo/MCM-41催化劑的HDS催化活性
圖5示出了Ni-Mo/MCM-41、Ni-Mo/TiO2-MCM-41和 TiO2-Ni-Mo/MCM-41催化DBT HDS反應的產物組成隨反應時間的變化。由圖5可以看出,DBT在Ni-Mo/MCM-41催化劑上HDS反應的主要產物為BP,其次為CHB,另外還有少量的芳環完全加氫產物BCH和加氫裂化產物苯和環己烷(B+C)。而對于 TiO2改性的Ni-Mo/MCM-41催化劑,DBT在產物中的含量有不同程度的降低,轉化率增加;從反應產物組成來看,DDS反應路徑的產物BP含量有所降低,CHB和BCH等加氫反應路徑產物尤其是BCH的量有了明顯增加;在較長反應時間下, CHB甚至取代了BP成為主要的反應產物。另外, DBT在 TiO2改性的 Ni-Mo/MCM-41催化劑上進行 HDS反應時,加氫裂化產物苯和環己烷的量也明顯提高。由于在有機硫化物如DBT存在的情況下,BP比較穩定,很難發生加氫裂化反應生成苯和/或環己烷[11],因此產物中苯和環己烷主要來源于加氫產物CHB和BCH的加氫裂化。從反應結果可以看出,TiO2的引入主要通過提高 Ni-Mo/ MCM-41催化劑 HYD反應路徑的活性提高了其HDS活性。比較圖5(2)和圖5(3)可以看出,將TiO2引入到Ni-Mo/MCM-41前驅體的表面對催化劑HYD和 HDS活性的促進作用更為明顯。
對于傳統的以γ-Al2O3作載體的雙金屬硫化物HDS催化劑,TiO2主要通過提高催化劑酸性[1]、提高活性組分分散度[2]、減弱活性組分與γ-Al2O3之間的相互作用等幾方面提高催化劑的 HDS反應活性[3]。另外,TiO2還起到了一定電子型助劑作用。在 HDS反應條件下,一部分 Ti4+可被部分還原為 Ti3+,Ti3+中3d軌道“富余”的電子注入 Mo的3d軌道后,使 Mo處于“富電子”狀態,削弱Mo—S鍵,產生更多的硫空穴或配位狀態不飽和的活性位,從而提高催化劑的活性[4]。但需要指出的是,目前廣泛用于描述DBT直接脫硫過程的機理主要有氫解[21]和β-消除[22]兩種。當催化劑表面處于富電子狀態時,對氫解和β-消除反應都有促進,主要提高催化劑的DDS反應路徑活性。這與本研究觀察到的結果,即 TiO2主要通過促進催化劑 HYD反應路徑的活性進而提高其 HDS反應活性不符。
根據 HDS反應產物的組成,TiO2改性的Ni-Mo/MCM-41催化劑表現出較高的加氫裂化活性,說明 TiO2的引入可能提高了Ni-Mo/MCM-41催化劑的酸性。但另一方面,圖4中 NH3-TPD結果表明,不同催化劑氧化態前驅體表面酸性質差別不大,因此推測 TiO2引入更有可能促進“金屬—SH”基團的生成,提高了硫化態催化劑的酸性。在酸性較強的催化劑上,活性中心能夠通過與酸中心的相互作用形成“缺電子結構(Electron-deficient structure)”[23],這部分具有缺電子性質的活性中心同時具有較高的加氫活性。另外提高催化劑的酸性,也有利于氫的解離和高活性溢流氫物種的生成,從而提高催化劑的加氫活性[24-25]。與 Ni-Mo/TiO2-MCM-41相比,在 TiO2-Ni-Mo/MCM-41中可能有更多的 Ti物種暴露在催化劑表面,能更為有效地提高硫化物催化劑的酸性,因此 TiO2-Ni-Mo/ MCM-41表現出更高的 HYD活性和加氫裂化活性。
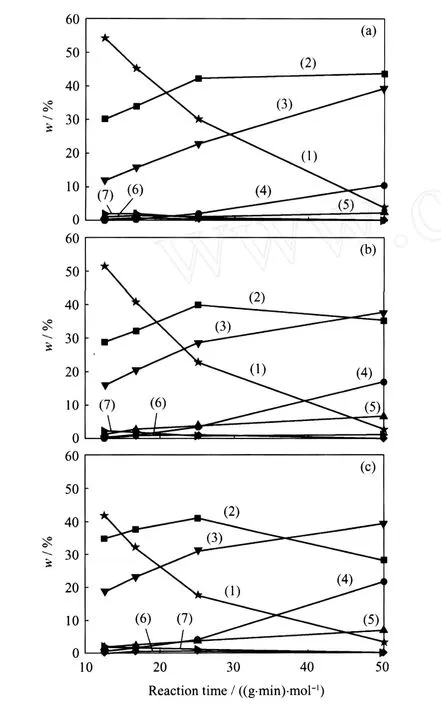
圖5 Ni-Mo/MCM-41、Ni-Mo/TiO2-MCM-41和TiO2-Ni-Mo/MCM-41催化DBT HDS反應的產物組成隨反應停留時間的變化Fig.5 Products compositions of DBT H DS over Ni-Mo/MCM-41, Ni-Mo/TiO2-MCM-41and TiO2-Ni-Mo/MCM-41 vs reaction time(a)Ni-Mo/MCM-41;(b)Ni-Mo/TiO2-MCM-41; (c)TiO2-Ni-Mo/MCM-41(1)DBT;(2)BP;(3)CHB;(4)B+C;(5)BCH; (6)HHDBT;(7)THDBTReaction time is specially appointed as the value of catalyst mass(g)/mol flow rate of reactant(mol/min)
3 結 論
(1)UV-Vis和 TPR結果表明,采用分步浸漬的方法將 TiO2先于 Ni-Mo活性組分引入對Ni-Mo/MCM-41前驅體中各物種的配位狀態影響不大,但促進了其前驅體中各物種的還原;反之則減少了催化劑前驅體中八面體配位的 Ni2+物種的含量,但是提高了八面體配位的 Mo6+物種的含量,并在一定程度上抑制了活性物種的還原。NH3-TPD結果表明,Ni-Mo/MCM-41、TiO2-Ni-Mo/MCM-41以及Ni-Mo/TiO2-MCM-41 3種催化劑氧化態前驅體表面以弱酸中心為主。但總的說來,TiO2的引入對Ni-Mo/MCM-41催化劑氧化物前驅體的配位狀態、還原性能以及酸性影響不顯著。
(2)TiO2的引入雖然抑制了 Ni-Mo/MCM-41的直接脫硫活性,但是顯著提高了其加氫反應路徑的活性,進而提高了Ni-Mo/MCM-41總的 HDS反應活性。其中,將 TiO2在Ni-Mo活性組分之后引入能更有效地提高 Ni-Mo/MCM-41催化劑活性。根據反應產物組成分析,TiO2可能主要通過提高Ni-Mo/MCM-41硫化物酸性提高了催化劑的 HYD和HDS活性。
[1]RAMíREZ J,RUíZ-RAMíREZ L,CEDENO L,et al. Titania-alumina mixed oxides as supports for molybdenum hydrotreating catalysts[J].Appl Catal:A, 1993,93(2):163-180.
[2]DAMYANOVA S,SPOJAKINA A,J IRATOVA K. Effect of mixed titania-alumina supports on the phase composition of NiMo/TiO2-Al2O3catalysts[J].Appl Catal:A,1995,125(2):257-269.
[3]鄧存,周振華,童迅.TiO2調變對 MoO3/γ-Al2O3和CoO-MoO3/γ-Al2O3催化性能的影響[J].分子催化, 1998,12(2):107-112.(DENG Cun,ZHOU Zhenhua, TONG Xun.Effect of TiO2modifiedγ-Al2O3on the catalytic behavior of MoO3/γ-Al2O3and CoO-MoO3/ γ-Al2O3[J].Journal of Molecular Catalysis(China), 1998,12(2):107-112.)
[4]RAMíREZA J,MACíASA G,CEDE?O L,et al.The role of titania in supported Mo,CoMo,NiMo and NiW hydrodesulfurization catalysts:An analysis of past and new evidences[J].Catalysis T oday,2004,98(1-2):19-30.
[5]YINGJ Y,MEHNERT C P,WONG M S.Synthesis and application of supramolecular templated mesoporous materials[J].Angew Chem Int Ed,1999,38(1-2):56-77.
[6]王瑤,王安杰,陳永英,等.以 MCM-41為載體擔載Ni-Mo硫化物制備柴油深度加氫脫硫催化劑[J].石油學報(石油加工),2003,19(5):36-41.(WANG Yao, WANG Anjie,CHEN Yongying,et al.Preparation of Ni-Mo sulfide catalyst supported by MCM-41 for diesel deep hydrodesulfurization[J]. Acta PetroleiSinica (Petroleum Processing Section),2003,19(5):36-41.)
[7]王瑤,王安杰,陳永英,等.以 MCM-41為載體擔載Co-Mo硫化物制備柴油深度加氫脫硫催化劑[J].石油學報(石油加工),2003,19(6):24-28.(WANG Yao, WANG Anjie,CHEN Yongying,et al.Preparation of Co-Mo sulfide catalyst supported by MCM-41 for diesel deep hydrodesulfurization[J]. Acta PetroleiSinica (Petroleum Processing Section),2003,19(6):24-28.)
[8]QIN W,ISHIHARA A,OGAWA S,et al.Study of hydrodesulfurization by the use of35S-labeled dibenzothiophene hydrodesulfurization mechanism on sulfided Mo/Al2O3[J].J Phys Chem,1994,98(3): 907-911.
[9]WANG A J,KABE T.Fine-tuning of pore size of MCM-41 by adjusting the initial p H of the synthesis mixture[J].Chem Commun,1999,20:2067-2068.
[10]HOUALLA M,NAG N K,SAPRE A V,et al. Hydrodesulfurization of dibenzothiophene catalyzed by sulfided CoO-MoO3/γ-Al2O3:The reaction network[J]. AICh E J,1978,24(6):1015-1021.
[11]REN J,WANG A J,LI X,et al.Hydrodesulfurization of dibenzothiophene catalyzed by Ni-Mo sulfides supported on a mixture of MCM-41 and HY zeolite[J]. Appl Catal:A,2008,344(1-2):175-182.
[12]ARNOLDY P,FRANKEN M C,SCHEFFER B,et al. Temperature-programmed reduction of CoO-MoO3/ A12O3catalysts[J].J Catal,1985,96(2):381-395.
[13]RAMíREZ J,CASTILLO P,CEDE?O L,et al.Effect of boron addition on the activity and selectivity of hydrotreating CoMo/Al2O3catalysts[J].Appl Catal: A,1995,132(2):317-334.
[14]建謀,楊先春,石亞華,等.氟在γ-Al2O3及 NiW/ γ-Al2O3催化劑中的作用 Ⅱ鎳和鎢的狀態[J].分子催化,1990,4(3):181-187.(J IAN Mou,YANG Xianchun,SHI Yahua,et al.Effects of fluoride inγ-Al2O3and NiW/γ-Al2O3catalysts Ⅱ The states of nickel and tungsten[J].Journal of Molecular Catalysis (China),1990,4(3):181-187.)
[15]HENKER M,WENDLANDT K P,VAL YON J,et al. Structure ofMoO3/Al2O3-SiO2catalysts[J]. Appl Catal,1991,69(1):205-220.
[16]RAMíREZJ,CONTRERAS R,CASTILLO P. Characterization and catalytic activity of CoMo HDS catalysts supported onalumina-MCM-41[J].Appl Catal:A,2000,197(1):69-78.
[17]BRITO JL,BARBOSA A L. Effectofphase composition of the oxidic precursor on the HDS activity of the sulfided molybdates of Fe(II),Co(II),and Ni(II) [J].J Catal,1997,171(2):467-475.
[18]RAN M S,MAITY S K,ANCHEYTA J,et al.TiO2-SiO2supported hydrotreating catalysts: Physicochemical characterization and activities[J].Appl Catal: A,2003,253(1):165-176.
[19]KLIMOVA T,RODRíGUEZ E,MARTíNEZ M,et al. Synthesis and characterization of hydrotreating Mo catalysts supported on titania-modified MCM-41[J].Microporous and Mesoporous Materials,2001,44-45:357-365.
[20]RYNKOWSKI J M,PARYJCZAK T,L ENIK M.On the nature of oxidic nickel phases in NiO/γ-Al2O3catalysts[J].Appl Catal:A,1993,106(1):73-82.
[21]TODOROVA T,PRINS R,WEBER T.A density functional theory study of the hydrogenolysis reaction of CH3SH to CH4on the catalytically active(100)edge of 2H-MoS2[J].J Catal,2005,236(2):190-204.
[22]BATAILL E F,L EMBERTEN J L,MICHAUD P, et al.Alkyldibenzothiophenes hydrodesulfurization——Promoter effect,reactivity and reaction mechanism[J]. J Catal,2000,191(2):409-422.
[23]ZENG S Q,BLANCHARD J,BREYSSE M,et al. Mesoporous materials from zeolite seeds as supports for nickel–tungsten sulfide active phases Part 2 Catalytic properties for deep hydrodesulfurization reactions[J]. Appl Catal:A,2006,298(1):88-93.
[24]AMBS W J,MITCHELL Jr M M.Hydrogen spillover on platinum-alumina,effect of water[J].J Catal,1983, 82(1):226-229.
[25]MILLER J T,MEYERS B L,MODICA F S,et al. Hydrogen temperature-programmed desorption (H2TPD)of supported platinum catalysts[J].J Catal, 1993,143(2):395-408.
猜你喜歡
課堂內外·初中版(科學少年)(2025年1期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2025年2期)2025-02-28 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
汽車觀察(2018年10期)2018-11-06 07:05:26
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
