延胡索總生物堿的提取純化工藝研究
2010-02-07 03:48:24王緒穎賈曉斌
中成藥 2010年11期
關鍵詞:工藝
王緒穎, 賈曉斌, 陳 彥
(1.江蘇省中醫藥研究院中藥新型給藥系統重點實驗室,國家中醫藥管理局中藥口服釋藥系統重點研究室,江蘇南京210028;2.江蘇大學藥學院,江蘇鎮江212013)
延胡索為罌粟科植物延胡索(Corydalis yanhusuo W.T.Wang)的干燥塊莖,主產地浙江,為著名的浙八味之一,具有活血化瘀、理氣止痛功效。現代藥理研究表明延胡索中主要有效組分延胡索總生物堿具有鎮痛、抗潰瘍、抑制胃酸分泌、解痙及增加冠脈血流量,抗心律失常等作用[1],總生物堿中延胡索乙素的鎮痛作用最強[2]。文獻中提取純化延胡索總生物堿,通常以延胡索乙素或總生物堿為指標[3-6],而同時以延胡索乙素和延胡索總生物堿為指標的則未見報道。
本實驗以延胡索乙素和總生物堿的含量為指標,以正交試驗優選延胡索總生物堿的乙醇回流提取方法,以延胡索乙素和延胡索總生物堿的比吸附量和比洗脫量為指標篩選出最佳純化樹脂,并考察其純化延胡索乙醇提取液的工藝條件及參數,為延胡索總生物堿的工業化生產提供切實的理論依據和條件。
1 儀器與材料
Agilent 1100高效液相色譜儀;十萬分之一電子天平(METTLER TOLEOR公司);UV-2802型紫外可見分光光度計(上海尤尼克儀器有限公司);DZF-6051真空干燥箱(上海精宏實驗設備有限公司);DK-S26型恒溫水浴鍋(上海精宏實驗設備有限公司)。
樹脂購自上海摩速科學器材有限公司;延胡索乙素對照品(批號:0726-200208,供含量測定用,由中國藥品生物制品檢定所提供);醋延胡索(批號:080201,南京藥業股份有限公司中藥飲片廠),經江蘇大學藥學院歐陽臻教授鑒定為罌粟科延胡索(Corydalis yanhusuo W.T.Wang)的醋制品;甲醇、磷酸為色譜純,水為超純水,其余試劑均為分析純。
2 方法與結果
2.1 延胡索乙素含量測定
2.1.1 色譜條件與系統適用性實驗 色譜柱Agilent HC-C18(150 mm × 4.6 mm,5 μm);流動相MeOH-0.1%H3PO4(三乙胺調節 pH=6)57 ∶43;流量:1.0 mL/min;檢測波長:280 nm。理論板數以延胡索乙素計不低于5 000,延胡索乙素色譜峰與相鄰未知色譜峰的分離度>1.5,色譜圖見圖1。
2.1.2 標準曲線的繪制 精密稱取延胡索乙素對照品3.42 mg至10 mL量瓶中,用流動相溶解并稀釋至刻度,搖勻,即得每1 mL含延胡索乙素0.342 mg的對照品貯備液;分別精密吸取貯備液0.1、0.2、0.4、0.8、1.0 mL 至 5 mL 量瓶,用流動相稀釋至刻度,搖勻;分別精密吸取上述對照品溶液20 μL進樣,測得峰面積積分值。以對照品溶液的進樣量為橫坐標,峰面積積分值為縱坐標,繪制標準曲線,得回歸方程 Y=45.674X+27.791,r=0.999 9,結果表明,延胡索乙素進樣量在0.136 8~1.368 μg與峰面積有良好的線性關系。
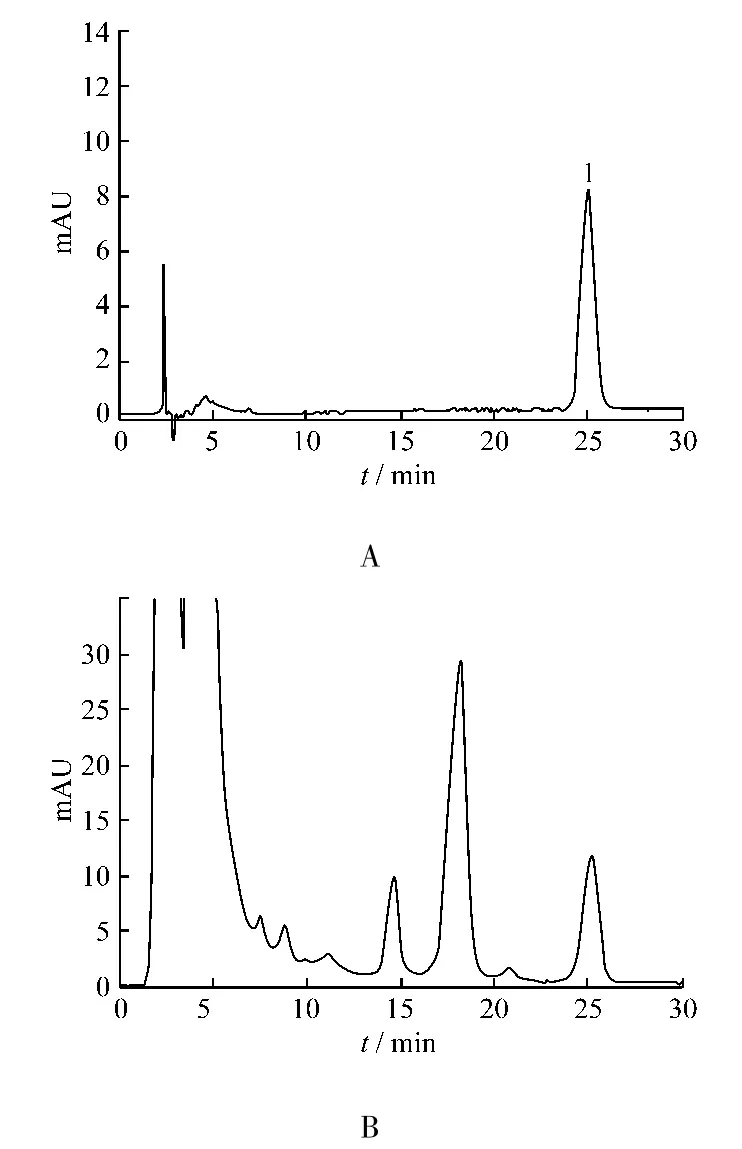
圖1 延胡索乙素對照品(A)與樣品(B)HPLC色譜圖,1為延胡索乙素
2.1.3 樣品的含量測定 吸取適量提取液或樹脂沖出液水浴揮干,用甲醇定容至5 mL,用0.45 μm的微孔濾膜濾過,取續濾液,精密吸取20 μL進樣,計算其含量。
2.2 延胡索總生物堿含量測定
2.2.1 對照品溶液制備 精密稱取延胡索乙素對照品2.22 mg至10 mL量瓶中,用pH4.5的乙酸-醋酸鈉緩沖液溶解并稀釋至刻度,搖勻,即得每1 mL含延胡索乙素0.222 mg的對照品溶液。
2.2.2 顯色方法及檢測波長的確定[7]精密吸取對照品溶液0.3 mL,至預先精密加入氯仿10 mL的分液漏斗中,再加0.7 mL pH4.5的醋酸-醋酸鈉緩沖液,精密加入4 mL溴甲酚綠溶液,振搖3 min,靜置40 min,分取氯仿層,用UV-2802型紫外可見分光光度計進行全波長掃描,在414 nm處有最大吸收峰,故選擇檢測波長為414 nm。
2.2.3 標準曲線的繪制 精密吸取對照品溶液0.0、0.1、0.2、0.3、0.4、0.5 mL,置預先精密加入氯仿10 mL的分液漏斗中,再分別加1.0、0.9、0.8、0.7、0.6、0.5 mL pH4.5 的醋酸-醋酸鈉緩沖液,精密加入4 mL溴甲酚綠溶液,振搖3 min,靜置40 min,分取氯仿層,在414 nm波長處測定吸光度,以對照品溶液的體積為橫坐標,吸光度為縱坐標,繪制標準曲線,得回歸方程為Y=60.798X-0.003 4,r=0.999 3,表明對照品濃度在0.002 22~0.011 1 mg/mL與吸光度之間呈良好的線性關系。
2.2.4 樣品的含量測定 取適量提取液或樹脂沖出液水浴揮干,用pH4.5的醋酸-醋酸鈉緩沖液定容至5 mL量瓶中,精密吸取1mL,按標準曲線項下同法操作,測定吸光度,計算其含量。
2.3 正交試驗優選延胡索的乙醇提取工藝
延胡索具有鎮痛止痛等功效,其主要藥效物質基礎生物堿類組分易溶于乙醇和酸水中,經過醋制后生物堿成鹽又能溶于水中,所以提取溶劑選擇含水乙醇。設計正交試驗進一步考察乙醇回流提取工藝,根據延胡索總生物堿的性質,以水、30%乙醇、50%乙醇、70%乙醇、90%乙醇為提取溶劑,進行平行性試驗,分別測定提取液中延胡索乙素和總生物堿的含量(mg/g生藥),試驗結果延胡素乙素含量分別為:0.115、0.232、0.419、0.308、0.197 mg/g生藥,總生物堿含量分別為:1.924、3.437、7.654、4.502、2.585 mg/g生藥,所以選擇30%、50%、70%乙醇作為正交試驗因素乙醇濃度的三個水平。對影響乙醇回流提取的主要影響因素:乙醇濃度、溶劑用量、回流提取時間和提取次數按四因素三水平表L9(34)進行正交試驗,各因素水平見表1,按正交試驗表安排試驗,平行2次,試驗結果見表2、表4,并對延胡索乙素和總生物堿的含量進行方差分析,結果見表3、表5。

表1 正交試驗因素水平
2.3.1 正交試驗樣品制備 分別精密稱取9份延胡索藥材,每份100 g,按正交試驗表各行各列條件制備樣品提取液,按含量測定項下制備樣品溶液。
2.3.2 正交試驗結果 以延胡索乙素含量為考察指標,由表2直觀分析表明影響延胡索乙素含量的主要因素為A,次要因素為D、C、B,方差分析表明A、D因素對延胡索乙素的含量有顯著意義(P<0.01),B、C因素無顯著意義,最佳組合為A2B3C3D3。以延胡索總生物堿含量為考察指標,表4直觀分析表明影響延胡索總生物堿含量的主要因素為A,次要因素為D、C、B,方差分析表明A因素對延胡索總生物堿的含量有顯著意義(P<0.01),B、C、D因素無顯著意義,最佳組合為A2B2C2D3。
不同考察指標下A、D的最佳條件為A2D3,B、C有差別,方差分析表明B、C無顯著意義,綜合生產實際,選擇B2C2,確定工藝條件為A2B2C2D3。

表2 正交試驗結果1(延胡索乙素含量指標n=2)
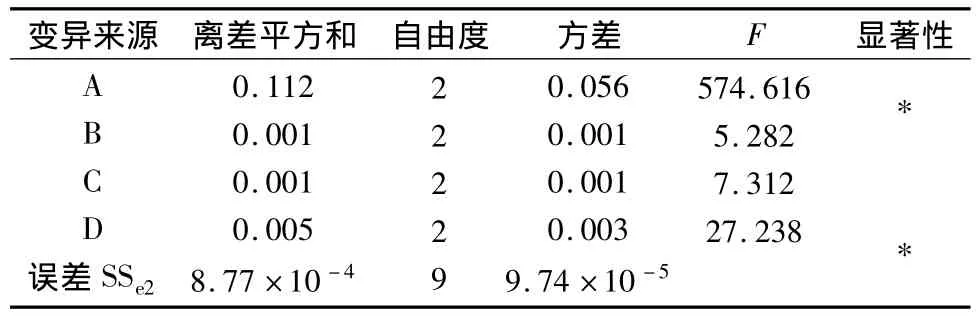
表3 延胡索乙素含量方差分析

表4 正交試驗結果2(延胡索總生物堿含量指標n=2)
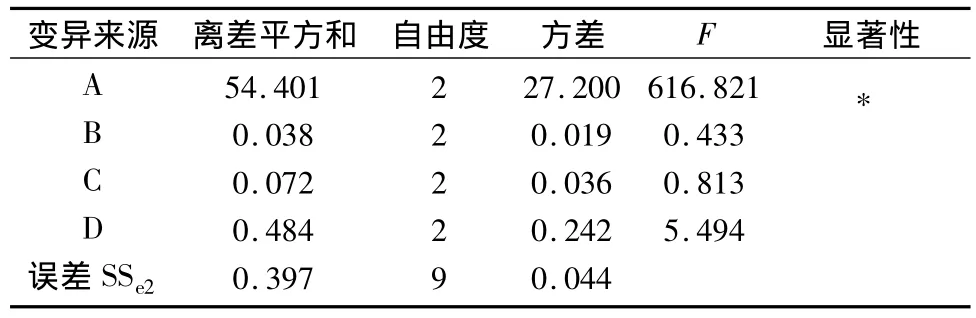
表5 延胡索總生物堿含量方差分析
2.3.3 驗證試驗 為進一步考察優選工藝的可靠性及穩定性,取3份藥材,每份1 000 g,用6倍量50%乙醇,加熱回流提取3次,每次2 h,然后分別測定各樣品中延胡索乙素和總生物堿含量,結果見表6。
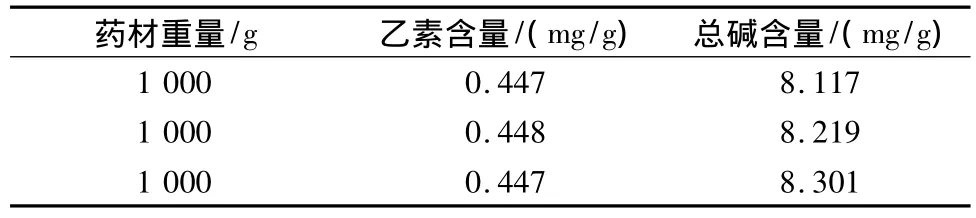
表6 驗證試驗結果
2.4 延胡索總生物堿純化工藝研究
延胡索生藥經正交試驗優化的最佳工藝提取后總生物堿的轉移率達85.6%,但得到的浸膏不容易干燥成粉,可能含較多水溶性多糖、果膠、黏液質及醇溶性油脂等,不利于制劑成型以及質量控制。本實驗以延胡索總生物堿和延胡索乙素含量為指標,用大孔吸附樹脂法對延胡索乙醇提取物進一步純化,篩選出延胡索生物堿的大孔樹脂分離富集工藝條件,為下一步制劑成型奠定技術基礎。
2.4.1 樣品制備 延胡索用6倍量50%乙醇,回流提取3次,每次2 h,合并濾液濃縮至1 g/mL,測得延胡索乙素含量為0.447 mg/g,延胡索總生物堿含量為8.212 mg/g。
2.4.2 樹脂預處理 先用2 mol/L NaOH浸泡5 h,用蒸餾水洗至中性。再用3 mol/L鹽酸浸泡8 h,用蒸餾水洗至中性。最后用95%乙醇浸泡24 h,充分溶脹,用95%乙醇洗至流出液加5倍水無白色渾濁,再用蒸餾水反復清洗至無醇氣味,用蒸餾水浸泡后待用。
2.4.3 樹脂型號確定 采用動態吸附法,比較四種樹脂對延胡索乙素和總生物堿的比吸附量、比洗脫量,優選出最佳樹脂。
稱取處理好的 AB-8、D101、HPD600 和 X-5 四種型號的濕樹脂各5.00 g,濕法裝柱,上樣53.00 mL,平行上樣3份,調節流速為0.5 mL/min,過柱流出液重吸附2次,靜置4 h,用蒸餾水洗至流出液無色,以70%乙醇洗脫10倍柱體積,分別用HPLC法和UV法測定流出液中延胡索乙素和總生物堿的含量,并分別計算比吸附量、比洗脫量,結果見表7、表8。
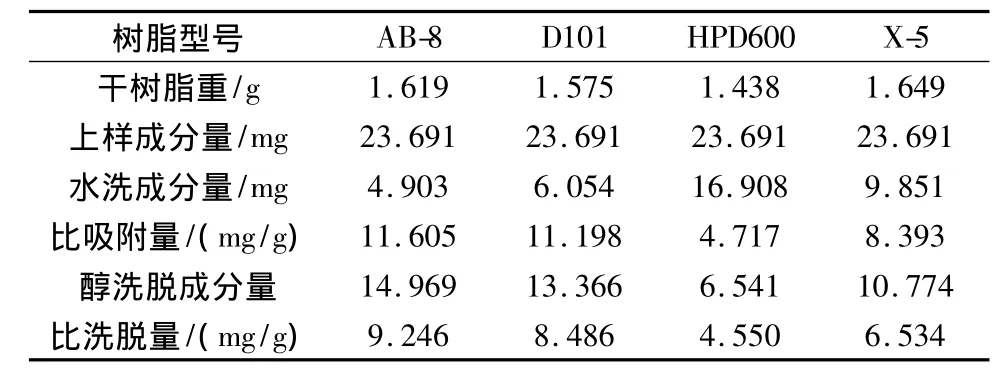
表7 4種型號大孔吸附樹脂對延胡索乙素的動態吸附性能比較(n=3)
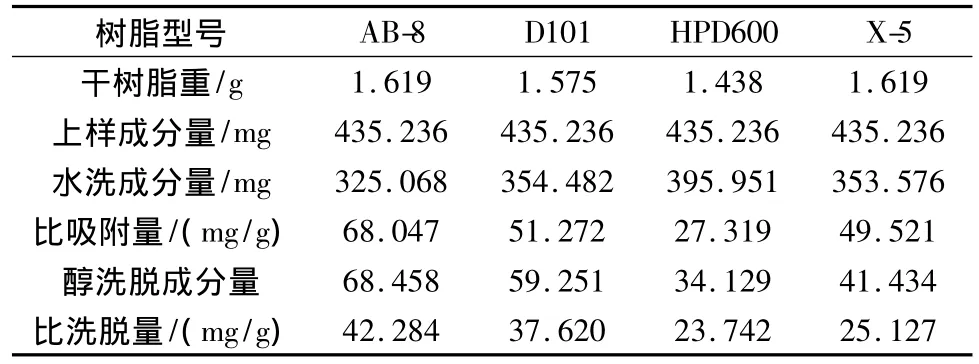
表8 4種型號大孔吸附樹脂對延胡索總生物堿的動態吸附性能比較(n=3)
由表7可知,樹脂AB-8對延胡索乙素的比吸附量為11.605 mg/g,10 BV70%乙醇比洗脫量為9.246 mg/g,均為4種樹脂中最大,折合生藥量每克干樹脂吸附25.962 g生藥。由表8可知,樹脂AB-8對延胡索總生物堿的比吸附量為68.047 mg/g,10BV70%乙醇比洗脫量為42.284 mg/g,均為4種樹脂中最大,折合生藥量每克干樹脂吸附8.286 g生藥。
不同考察指標下均篩選出AB-8為最佳樹脂,但不同考察指標下樹脂對生藥的比吸附量不同,其中延胡索乙素折合生藥的樹脂比吸附量遠大于延胡索總生物堿折合生藥的樹脂比吸附量,為盡量保留有效組分,選擇以延胡索總生物堿為指標下的飽和吸附量作為生藥的最佳吸附量,即8.286 g/g。
2.4.4 水洗體積確定 按延胡索總生物堿的飽和吸附量上樣,精密吸取樣品液(1 g/mL)8.29 mL加至已處理好的裝有5 g濕AB-8大孔吸附樹脂柱,調節流速為0.5 mL/min,過柱流出液重吸附2次,靜置4 h,用蒸餾水洗脫,每1 BV收集在一起,分別用Molish反應檢測,第6 BVMolish反應陰性,證明不吸附的雜質已基本洗脫完全,確定上柱后水洗體積為6 BV。
2.4.5 洗脫溶劑濃度確定 按延胡索總生物堿的飽和吸附量上樣,精密吸取樣品液(1 g/mL)8.29 mL加至已處理好的裝有5 g濕AB-8大孔吸附樹脂柱,調節流速為0.5 mL/min,過柱流出液重吸附2次,靜置4 h,用蒸餾水洗脫至流出液無色,依次用40%乙醇、50%乙醇、60%乙醇、70%乙醇、80%乙醇、90%乙醇各10 BV洗脫,洗脫液流速為1.0 mL/min,測定洗脫液中延胡索乙素和總生物堿的量并分別計算洗脫百分率,結果見表9。

表9 洗脫溶劑濃度的確定
由表9可知,隨著乙醇體積分數的增加,AB-8樹脂對延胡索乙素和延胡索總生物堿的累積洗脫率逐漸增大,但90%乙醇洗脫的量增加的很少,綜合生產實際選擇80%乙醇作為洗脫溶劑。
2.4.6 洗脫溶劑體積確定 按延胡索總生物堿的飽和吸附量上樣,精密吸取樣品液(1 g/mL)8.29 mL加至已處理好的裝有5 g濕AB-8大孔吸附樹脂柱,調節流速為0.5 mL/min,過柱流出液重吸附2次,靜置4 h,用蒸餾水洗脫6倍柱體積,用80%乙醇洗脫,按 1 BV、2 BV、3 BV、4 BV、5 BV、6 BV、7 BV、8 BV、9 BV、10 BV、11 BV、12 BV、13 BV、14 BV、15 BV、16 BV進行收集,繪制洗脫曲線。分別測定延胡索乙素和總生物堿的含量,并分別以延胡索乙素和總生物堿的比洗脫量為縱坐標(mg/g),洗脫柱體積(BV)為橫坐標繪制延胡索乙素和總生物堿的洗脫曲線,見圖2。
由圖2可知,第11 BV80%的乙醇已經基本洗脫不到延胡索乙素和總生物堿,所以選擇10 BV作為洗脫溶劑的最佳洗脫體積。
2.4.7 驗證試驗 為進一步考察優選工藝的可靠性及穩定性,吸取82.86 mL樣品液,按上述確定的最佳工藝條件,加至已處理好的裝有50 g濕AB-8大孔吸附樹脂柱,調節流速為0.5 mL/min,過柱流出液重吸附2次,靜置2 h,用蒸餾水洗脫6倍柱體積,用80%乙醇洗脫10 BV,收集乙醇洗脫液,回收乙醇,80℃真空干燥。測定延胡索乙素和延胡索總生物堿含量及各自占干膏的比例。結果見表10。
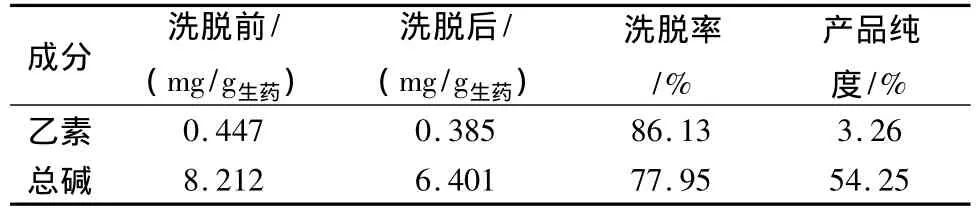
表10 驗證試驗結果
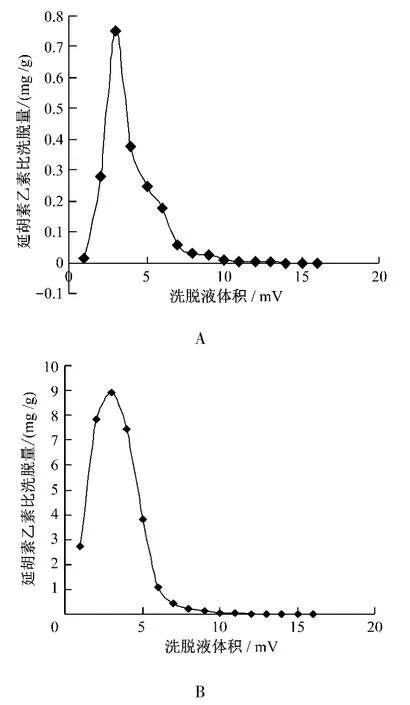
圖2 80%乙醇洗脫曲線,A為延胡索乙素指標,B為延胡索總生物堿指標
3 結論與討論
乙醇回流提取延胡索的最佳工藝條件是6倍量50%乙醇,加熱回流提取3次,每次2 h;AB-8樹脂純化延胡索乙醇提取液的最佳工藝條件是每克干樹脂吸附8.286 g生藥,上柱后水洗6 BV,80%乙醇洗脫10 BV。該工藝經驗證,總生物堿的提取轉移率達85.6%,純化轉移率達77.95%,終產品總生物堿純度為54.25%,該工藝穩定可靠,可較好的提取純化延胡索中總生物堿。
在篩選上柱液最佳pH的試驗中,發現將上柱液pH調為堿性時有沉淀析出,且堿性越強,產生沉淀越多,易堵塞樹脂柱,不利于樹脂柱的吸附。pH調為酸性時雖然比吸附量增大,但比洗脫量下降,而因為實驗所用的延胡索為醋制,上柱原液pH為5.20,比吸附量和比洗脫量都較大,所以上柱液最佳pH確定為原液pH。
[1]黃泰康.常用中藥成分與藥理手冊[M].北京:中國醫藥科技出版社,1994:874.
[2]馬勝興,陳可冀.延胡索研究概況[J].中西醫結合雜志,1985,5(12):758.
[3]胡季強,王如偉,邱紅英,等.大孔樹脂分離純化延胡索總生物堿的工藝研究[J].中國現代應用藥學雜志,2009,26(4):271-273.
[4]盧耀文,楊中林.延胡索乙素提取工藝優選研究[J].中成藥,2008,30(8):1247-附1.
[5]黃諾嘉.珍杉理胃片中延胡索生物堿提取工藝的研究[J].中成藥,1999,21(12):622-623.
[6]劉俊紅,魏峻峰,王洪志,等.大孔吸附樹脂在延胡索生物堿提取分離中的應用[J]. 中草藥,2002,33(1):73-83.
[7]周毅玲.酸性染料比色法測定延胡索總生物堿的量[J].中草藥,2008,39(8):1257-1258.
猜你喜歡
中國特種設備安全(2022年5期)2022-08-26 09:19:32
礦產綜合利用(2020年1期)2020-07-24 08:50:40
山東冶金(2019年6期)2020-01-06 07:45:54
收藏界(2019年2期)2019-10-12 08:26:06
世界農藥(2019年2期)2019-07-13 05:55:12
世界農藥(2019年2期)2019-07-13 05:55:10
模具制造(2019年3期)2019-06-06 02:11:00
山東工業技術(2016年15期)2016-12-01 05:30:59
銅業工程(2015年4期)2015-12-29 02:48:39
新疆鋼鐵(2015年3期)2015-11-08 01:59:52
