微乳薄層色譜分離鑒別百葉消炎洗劑主要成分
2010-02-07 03:48:24郎軼詠朱曉紅劉蘭娣姜同英
中成藥 2010年11期
郎軼詠, 王 強, 朱曉紅, 劉蘭娣, 姜同英
(1.中國人民解放軍第二0二醫院藥劑科,遼寧沈陽1100032;2.沈陽藥科大學藥學院,沈陽110015)
百葉消炎洗劑為中國人民解放軍第二〇二醫院院內制劑,具有清熱解毒,祛風燥濕,殺蟲止癢的功效。用于婦女陰道炎,附件炎,赤帶白下,淋病,尖銳濕疣等男女生殖器官疾病及痔瘡,腳氣等癥狀。百葉消炎洗劑由敗醬草,土茯苓,苦參,夏枯草,蛇床子等十多味中藥經過提取加工制成,成分復雜,分離比較困難,至今還沒有統一的鑒別標準。中藥成分的鑒別常采用TLC法,經典TLC法鑒別時展開劑常為多種有機溶劑,樣品的前處理也比較繁瑣,為了簡化鑒別步驟,節省鑒別時間,筆者以含微乳液作為展開劑,聚酰胺薄膜為固定相,研究了百葉消炎洗劑的微乳薄層色譜行為,建立了微乳薄層色譜(ME-TLC)鑒別百葉消炎洗劑的有效成分(夏枯草、敗醬草、蛇床子),取得了滿意的結果[1]。
1 儀器與試藥
2F-1三用紫外分析儀(上海精科實業有限公司);層析用聚酰胺薄膜(規格8 cm×20 cm,浙江省臺州是路橋四甲生化塑料廠)。百葉消炎洗劑(解放軍第202醫院,批號:20080824);對照藥材:敗醬草,夏枯草,蛇床子,由202醫院制劑室提供;十二烷基硫酸鈉(SDS)(湖南爾康制藥有限公司);其它試劑均為分析純。
2 方法
2.1 微乳層析液的制備
取SDS,精密稱定,溶解于適量水中,加入油相、助表面活性劑,加水至足量,磁力恒溫攪拌混勻,放置24 h即得澄清透明的微乳液。將各種改性劑加至組成不同的微乳液中,即得組成不同的微乳層析液。
2.2 供試品溶液的制備
取百葉消炎洗劑50 mL,加入95%乙醇醇沉2次,過濾,濃縮作為供試品溶液。
2.3 對照藥材溶液的制備
分別取敗醬草、夏枯草、蛇床子、對照藥材適量,加水浸泡24 h后,加熱至煮沸后在60℃ 條件下加熱煎煮3 h,收集煎出液,過濾,加與水煎液等體積的95%乙醇醇沉2次,放置0.5 h,濾過,合并濾液,濃縮,作為對照藥材溶液。
2.4 陰性對照溶液的配制
按處方比例及工藝,分別制備敗醬草,夏枯草,蛇床子陰性對照溶液。
2.5 微乳薄層色譜
分別吸取敗醬草、夏枯草、蛇床子的對照藥材溶液、陰性對照液及百葉消炎洗劑的供試品溶液各2 μL,點于聚酰胺薄膜,直徑為1~2 mm。以微乳層析液為展開劑,上行法展開,展程約11 cm,取出晾干后,置紫外光(365 nm)下檢視,對照品溶液與供試品溶液在相同位置顯現相同顏色的熒光斑點。
3 結果
3.1 微乳展開體系的選擇
3.1.1 微乳展開劑含水量對展開效果及時間的影響
在保證SDS-正丁醇-正庚烷比例為0.27 ∶0.63 ∶0.10(w/w)的條件下,配制系列含水量不同的微乳液展開劑。以微乳液-甲酸-丙酮(4∶1∶1)為展開劑進行微乳薄層層析,測定含水量對百葉消炎洗劑主要成分斑點遷移的影響,以及其在不同含水量的微乳液中層析10 cm距離所需要的時間。結果見圖1,2。
百葉消炎洗中的敗醬草、夏枯草、蛇床子斑點的Rf值隨著微乳液含水量增加而逐漸降低(見圖1)。微乳液的含水量對層析時間有顯著的影響,含水量越低,層析10 cm距離所需的時間越長。含水量在15% ~75%之間時(見圖2),3種主要成分相互之間的△Rf值隨著微乳液含水量的增加而顯著變大,且當水分含量為75%時達到最大值;當含水量大于75%時,△Rf隨著微乳液含水量的增加而降低。當水分含量小于30%時,微乳類型為W/O型,各主要成分相互間的△Rf值較小,尤其是敗醬草和蛇床子的斑點有部分重疊且不規則,沒能徹底分離,分離效果不理想,且展開時間較長(>6 h)。在W/O~O/W型(含水量45% ~60%)微乳液中,展開時間縮短,分離效果雖然得到改善,斑點基本都能分離,但斑點仍有擴散現象,且個別△Rf值略有降低,分離效果不夠理想。在O/W型(含水量>75%)微乳液中,分離效果得到顯著的改善,但當含水量過大,大于90%,由于展開劑極性太大,薄層前沿分層明顯,斑點略有拖尾現象,且分離度也降低。因此結合斑點效果和分離度以及時間等因素綜合考慮,選擇含水量75%的O/W型微乳液作為展開劑最為適宜。
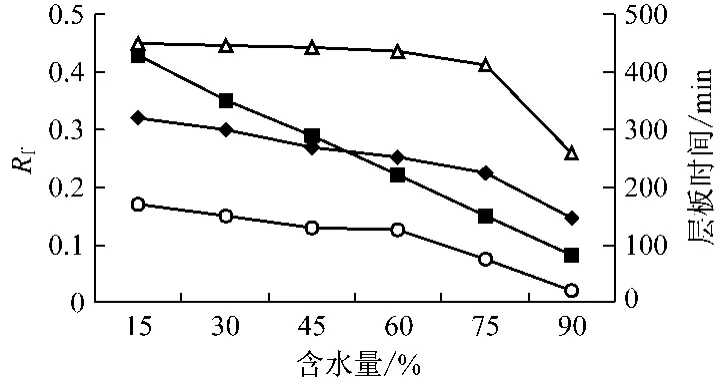
圖1 百葉各主要成分Rf值、時間與含水量的關系圖
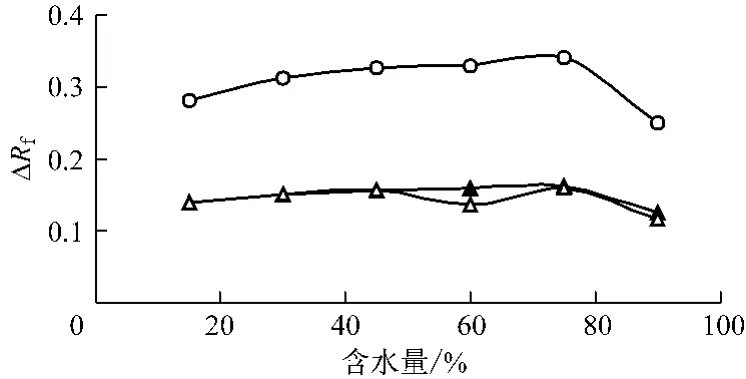
圖2 百葉主要各成分△Rf與含水量的關系
3.1.2 油相對展開效果的影響
保證微乳液(含水量75%,其它同3.1.1項組成)組分不變的情況下,改變油相的種類,分別考察了正戊烷、正己烷、正庚烷和正辛烷4種油相對展開效果的影響。見表1。
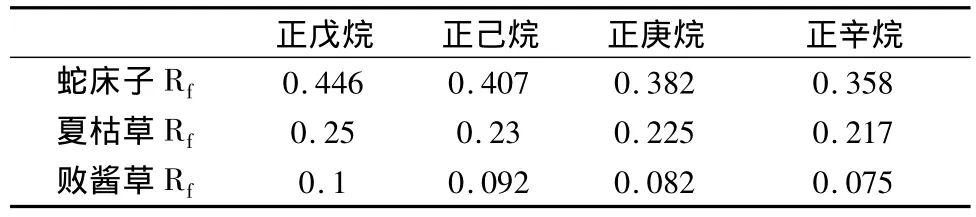
表1 百葉各主要成分Rf值與油相種類的關系
敗醬草、夏枯草和蛇床子的Rf值隨油相碳連數增加而逐漸減小。可解釋為:大分子油相不易嵌入表面活性劑中,而小分子油相可以像助表面活性劑一樣容易嵌入表面活性劑中形成界面膜,即油相分子體積越小,對藥物的溶解力越強,因此,為了增加藥物的溶解度,增大微乳形成的區域,應該選擇短鏈油相。當百葉薄層色譜中使用正辛烷時,由于正辛烷的碳鏈長,極性大,對藥物的溶解力小,從而使各藥物的Rf值降低,分離不完全,因此,應該選擇短鏈油相-正戊烷,正己烷和正庚烷,但是正戊烷和正己烷的層析結果顯示擴散和拖尾現象嚴重,因此,選擇正庚烷為微乳液的油相。
3.1.3 SDS對展開效果的影響
微乳薄層色譜常用的表面活性劑為十二烷基硫酸鈉(SDS)[1],分別取不同量的SDS 與15.8 g 正丁醇、2.5 g 正庚烷及75.0 g重蒸餾水配成展開劑,薄層展開,各對照藥材主斑點Rf值見表2。
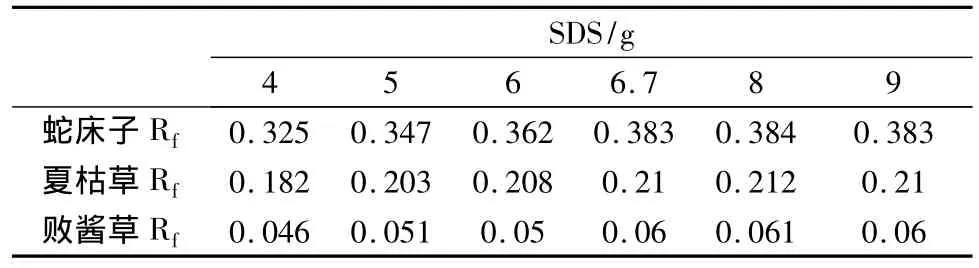
表2 百葉各對照藥材主斑點Rf值與SDS量的關系
敗醬草、夏枯草和蛇床子主斑點Rf值隨SDS量的增加先逐漸增大(見表2),SDS量大于6.7 g后,變化逐漸減小。且層析結果表明SDS為6.7 g時,各斑點的分離度最好。當SDS濃度較低時,微乳液滴的增溶能力較弱,對脂溶性較大的成分增溶作用小,隨著表面活性劑濃度的增加,對脂溶性成分的增溶作用增強,微乳分離能力提高,被分離成分間比移值增大。而當SDS濃度達到6.7 g,在微乳其他成分不變的情況下,對各主要組分比移值的影響并不明顯。同時隨著SDS濃度的增加,微乳液滴增大,展開時間延長。綜合考慮選擇SDS為6.7 g微乳液作為展開劑最為適宜。
3.1.4 助表面活性劑種類對分離效果的影響
微乳薄層色譜法中常加入助表面活性劑改善分離效果,本試驗考察了正丁醇、異丙醇、乙醇、甲醇4種助表面活性劑對展開效果的影響,結果見表3。異丙醇不能與微乳液很好的相溶,微乳液分層,不能作為助表面活性劑。所以,助表面活性劑的種類實驗最后只有正丁醇、甲醇和乙醇。
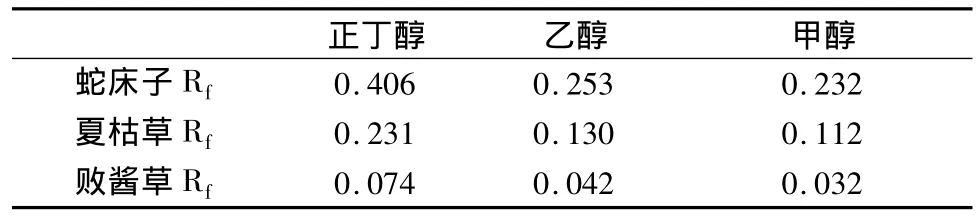
表3 助表面活性劑種類對分離效果的影響
助表面活性劑作為微乳系統中的重要組成部分,由于其分子處于膠束的表面活性劑分子之間,降低了它們的相互排斥,因此可以降低表面張力使微乳系統保持穩定。由表3可以看出,助表面活性劑的種類對分離效果的影響較大。以甲醇或乙醇代替正丁醇后,百葉各成分的Rf都明顯減小,且各物質的分離度都降低,分離效果不佳。這是由于碳鏈較短的助表面活性劑被吸附入表面活性劑極性端的一側,碳鏈較長的助表面活性劑則嵌入表面活性劑的碳鏈中[2]與非極性物質競爭表面活性劑分子上的非極性基團,使得表面活性劑對非極性物質的作用力減弱,非極性物質的Rf值增加。所以,選用正丁醇作為助表面活性劑最為適合。
3.2 改良劑對層析效果的影響
在保證展開劑SDS-正丁醇-正庚烷(0.27 ∶0.6 ∶0.10)+75%水組分不變的情況下,考察了多種改性劑,如甲酸,乙酸,丙酮,乙酸乙酯,氯仿,氨水等對展開效果的影響。結果表明乙酸乙酯,氯仿,丙酮加入后不能形成微乳。乙酸、氯仿的加入不能改善層析結果中的拖尾現象。甲酸的加入可明顯改善斑點的拖尾現象。在此基礎上考察了甲酸用量對展開效果的影響結果見圖3~4。
由圖3可以看出:在不含甲酸的微乳液展開劑中,有少量斑點拖尾,對分離造成不良影響,當甲酸/微乳液比例大于1/7時,敗醬草、夏枯草和蛇床子的Rf值隨著甲酸比例的增加而增加,并且斑點的拖尾現象逐漸改善。故甲酸加入量>1/7即可達到效果,但從圖可看出當甲酸加入量達到1/3夏枯草的Rf值略有下降,這可能是由于甲酸用量過多破壞了微乳液的穩定性的緣故。因此,選擇加入1/4甲酸作為改性劑。
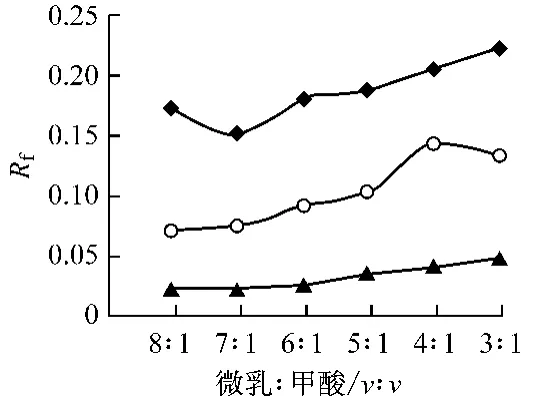
圖3 百葉各成分的Rf值與微乳液/甲酸比例變化關系圖
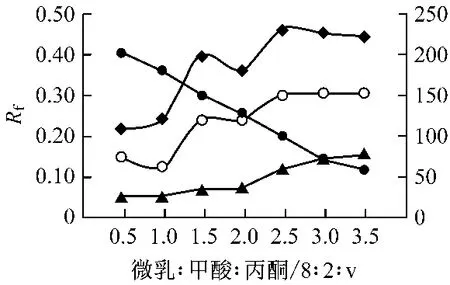
圖4 百葉各成分的Rf值與微乳液/甲酸/丙酮比例的變化的關系圖
實驗中發現丙酮的加入可明顯縮短展開時間且不影響展開效果,因此,選擇甲酸、丙酮同時作為改性劑,以獲得最佳層析效果。丙酮加入量對展開效果的影響見圖4。敗醬草的Rf值隨著丙酮含量的增加而增大,當微乳液-甲酸-丙酮比例達到8∶2∶2后,比移值幾乎沒有變化;夏枯草的Rf值隨著丙酮量的增加而逐漸增大,當微乳液-甲酸-丙酮比例達到8∶2∶2后,Rf值的增大幅度減少。蛇床子的Rf值也呈增大趨勢,當微乳液-甲酸-丙酮比例達到8 ∶2 ∶2后,Rf值減小。并且隨著丙酮的加入各主要成分的分離度逐漸增大。但丙酮量過多時可使展開時間過短,導致各成分不能得到充分的展開,分離效果不好。因此,由上圖可看出微乳液-甲酸-丙酮比例為8∶2∶2時效果最佳。綜上所述,在同時加入甲酸、丙酮為改性劑時可使分離效果得到顯著改善。并且可以縮短層析時間。因此,選擇含水量為75%微乳液-甲酸-丙酮比例為8∶2∶2的層析液為展開劑。
3.3 層析結果
含水量75%微乳液-甲酸-丙酮(4∶1 ∶1)為展開系統百葉消炎洗劑薄層色譜見圖5。
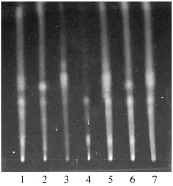
圖5 百葉消炎洗劑薄層色譜圖
4 討論
微乳薄層色譜的原理根據膠束色譜理論[2-3]大致相同,當以微乳為流動相時,溶質的分配可能是在固定相、微乳液的水連續相及油核和界面膜數相之間進行。由于微乳液的富集增溶,降低界面張力作用,極性大的分子分配于微乳水連續相中,極性小的分子分配于微乳油核或穿插排列于表面活性劑與助表面活性劑組成的膜柵欄中。并且由于分配、吸附、靜電、疏水、立體等可能的效應,使待測藥品中個組分在微乳液中展開是遷移速度不同,使其具有獨特的選擇性,可同時分離親水物質和疏水物質,對帶電成分和非帶電成分亦有較好的分離選擇,適用于分離結構和性質差別細微的復雜組分的物質,且更有利于提高色譜效率。中成藥的成分復雜,有效成分的鑒定比較困難。本課題研究了微乳薄層色譜法對百葉消炎洗劑有效成分的分離鑒定。與常用展開劑相比,以微乳液作為展開劑,被測組分與其它組分的分離度顯著提高,樣品中檢出的斑點明顯增多;與對照品對應的各斑點清晰、圓而集中,即使靠近前沿,斑點也不擴散;熒光強度增強,提高了檢測的靈敏度。由于一次檢出組分較多,展距增加到12 cm左右,可使分離效果更加理想。本研究同時對同一藥物中幾種有效成分在微乳中的色譜行為進行探討。實驗表明,性質差別較大的各類組分(如夏枯草和敗醬草)和性質結構差別細微的同系物可同時進行分離鑒定,為中成藥有效成分的分離鑒定提供了一種高效簡捷的新方法[4]。
[1]康 純,聞璃毓,丁仲伯.微乳薄層色譜用于黃酮類藥物分離鑒定的研究[J].藥物分析雜志,2000,20(2):121.
[2]張正全,陸 彬.微乳給藥系統研究概況[J].中國醫藥工業雜志,2001,32(3):139.
[3]戈早川,林輝概,李志良.膠束色譜與包合色譜的概況與進展[J].分析化學,1991,19(9):1092.
[4]郭 榮,朱霞石.微乳液介質中硫氰酸鐵的分光光度測定[J].高等學校化學學報,1987,8(6):508.
