慢病毒載體法制備紅色熒光蛋白轉基因小鼠
2010-02-16 14:55:36賈俊雙肖高芳林曉琳姚志芳杜文婷申紅芬饒子亮姚開泰
中國比較醫學雜志 2010年2期
關鍵詞:小鼠
賈俊雙,肖高芳,林曉琳,張 晟,姚志芳,杜文婷,申紅芬,劉 科,饒子亮,孫 妍,姚開泰,肖 東,
(南方醫科大學1.腫瘤研究所,2.比較醫學研究所暨實驗動物中心,廣州 510515;3.廣東省醫學實驗動物中心,廣州 528248)
轉基因小鼠已成為研究基因功能、開展發育生物學研究、建造人類疾病動物模型及研究人類疾病發病機制等的最重要的和最佳的模式實驗動物[1]。原核顯微注射法為目前最經典的和最成熟制備轉基因小鼠的方法,但其制備轉基因動物的效率很低。近年來,隨著不同用途慢病毒載體的問世,慢病毒介導的轉基因動物制作方法逐漸受到人們青睞;與原核顯微注射法相比,慢病毒載體法主要優勢如下:(1)操作較原核顯微注射法簡單的多,容易被一般人掌握;(2)對用于制作轉基因小鼠的小鼠品系無特殊要求,原核顯微注射時注射針必須插入核膜清晰的核內,因不同品系小鼠原核大小和核膜清晰度差異明顯,而慢病毒受精卵卵周隙注射時,注射針無需插入核內;(3)高的胚胎存活率,因慢病毒卵周隙注射時對細胞膜和核膜均無損傷;(4)慢病毒載體法的轉基因效率遠高于原核顯微注射法[2,3]。
利用遺傳工程小鼠在體內解析基因功能的策略可分為:基因功能缺失(Loss of gene function)和基因功能獲得(gain of gene function);在體內實現基因功能缺失的策略主要包括:(條件性)基因敲除、(條件性)轉基因RNA干涉(RNAi)和負顯性突變體(dominant negative mutant)技術等,而在體內實現基因功能獲得的主要策略:目的基因(條件性)過表達等;近若干年來,針對不同基因采用不同的基因功能研究策略,利用不同類型的慢病毒載體借助慢病毒載體法建立了相應的轉基因小鼠,在轉基因小鼠體內已實現或有望實現如下目標:(1)基因功能獲得[目的基因(條件性)過表達]和(2)基因功能缺失[(條件性)轉基因RNAi和負顯性突變體技術],以上策略和方法許多已成功用于在小鼠體內解析基因功能,這些都說明基于慢病毒載體法制備的轉基因小鼠為活體內研究基因功能提供了理想三維研究體系[2-10]。
1 材料和方法
1.1 材料
1.1.1 載體和細胞:慢病毒載體phUb-mRFP[11]由美國斯坦福大學的Sanjiv Sam Gambhir博士惠贈,phUb-mRFP中含紅色熒光蛋白(monomeric red fluorescence protein,mRFP)基因,其表達受人泛素啟動子(human ubiquitin promoter,hUb promoter)調控。慢病毒包裝系統購自Invitrogen公司。293FT細胞來源和培養方法等參見文獻[12]。
1.1.2 實驗動物:ICR小鼠(SPF級,6~8周齡,質量16~22g)由上海斯萊克實驗動物中心提供(許可證號:SCXK(滬)2007-0005),飼養于SPF級屏障系統內,設施使用合格證號:SYXK(粵)2006-0074。ICR小鼠用作以下群體:供受精卵用雌鼠、種公鼠、假孕母鼠和結扎雄鼠。
1.1.3 主要試劑:PCR擴增試劑dNTP和Taq酶及限制性內切酶等購自大連TaKaRa公司,質粒提取試劑盒購自QIAGEN公司,KSOM培養液和FHM操作液購自Chemicon公司,透明質酸酶、礦物油和細胞松弛素購自Sigma公司。細胞培養和轉染用試劑等來源參見文獻[12],其余試劑為國產或進口化學純或分析純。
1.2 方法
1.2.1 phUb-mRFP酶切鑒定:用NotI、MluI和BamHI對phUb-mRFP進行單酶切,酶切產物行瓊脂糖凝膠電泳。
1.2.2 慢病毒的包裝、濃縮及鑒定:慢病毒包裝、濃縮和保存步驟和方法詳見文獻[12]和Invitrogen公司操作手冊。經超速離心濃縮的病毒分裝后于-80℃保存備用;用濃縮后的病毒感染293FT細胞,24~48h后熒光顯微鏡下觀察是否見紅色熒光,以確認病毒是否成功生產。
1.2.3 慢病毒卵周隙注射制備mRFP轉基因小鼠:超數排卵后采集受精卵,然后將濃縮后的病毒注射入小鼠受精卵的卵周隙中,并將注射后狀態良好的受精卵移植進ICR假孕母鼠輸卵管內,仔鼠一般19.5~20.0d后出生。假孕母鼠和結扎雄鼠制備以及超數排卵、受精卵采集、病毒卵周隙注射和受精卵移植等方法參見文獻[3,13]。
1.2.4 利用體視熒光顯微鏡篩選與鑒定mRFP轉基因首建鼠:將出生幾天后的仔代乳鼠置于體視熒光顯微鏡下,檢測mRFP表達以初篩mRFP轉基因鼠,并從中選出熒光強度適中的乳鼠,作標記。此外,小鼠出生3周后,剪取6只F0代鼠的鼠耳,并置于體視熒光顯微鏡下,檢測mRFP表達以進一步鑒定mRFP轉基因鼠,并從中選出熒光強度適中的mRFP轉基因首建鼠。
1.2.5 采用PCR進行基因型鑒定以驗證體視熒光顯微鏡鑒定結果:在利用體視熒光顯微鏡初篩和鑒定mRFP轉基因鼠的基礎上,應用PCR驗證mRFP陽性的轉基因鼠基因組是否成功整合轉基因mRFP。步驟與方法如下:
1.2.5.1 鼠尾基因組DNA提取:小鼠出生3周后,剪鼠尾,采用基因組DNA提取試劑盒(申能博彩生物科技有限公司)從潛在的mRFP轉基因鼠和野生型ICR鼠(陰性對照)鼠尾組織提取基因組DNA,操作參見試劑盒說明書和文獻[14,15]。
1.2.5.2 基因型鑒定PCR擴增:PCR擴增轉基因用引物:P1,5′-CGATGACTTACTGGCGGGT-3′;P2,5′-CGCACCGTATTGGCAAGCA-3′。PCR反應條件:預變性95℃7min;變性95℃50s,退火54℃50s,延伸72℃1min,30個循環;延伸72℃,7min。PCR擴增后,取5μL反應液進行2%瓊脂糖凝膠電泳。
1.2.6 mRFP轉基因首建鼠繁殖傳代及轉基因遺傳和表達穩定性檢測:將mRFP表達陽性及PCR陽性的首建鼠(R3和R4)與野生型ICR鼠交配以傳代,獲得F1后,用小動物活體成像儀檢測mRFP是否在F1代表達,對其表達穩定性作出判斷,并進而間接判斷外源轉基因是否穩定遺傳。
2 結果與分析
2.1 慢病毒載體phUb-mRFP鑒定
phUb-mRFP經NotI、MluI和BamHI分別單酶切,產物經電泳均可見兩條帶,其大小與理論預測值相符(圖1)。此外,對phUb-mRFP載體啟動子進行測序亦證實phUb-mRFP載體是正確的(數據未顯示)。
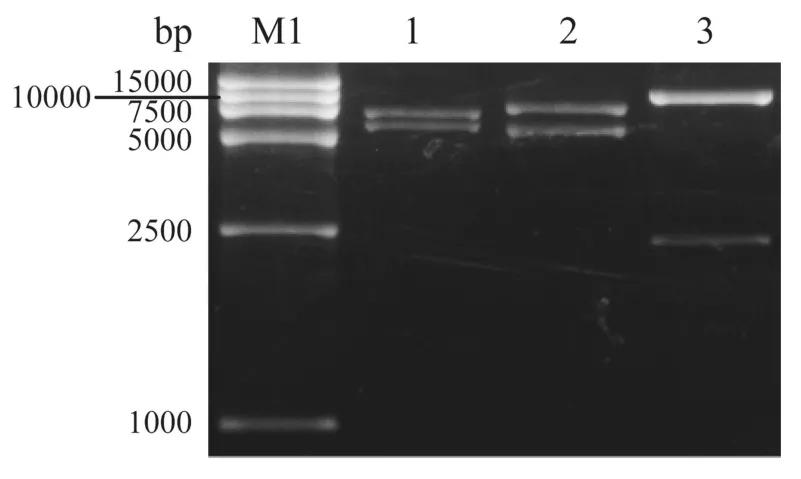
泳道1:DL15 000 marker(TaKaRa);泳道1~3:NotI,MluI或BamHI分別酶切phUb-mRFP圖1 慢病毒載體phUb-mRFP酶切鑒定LaneM1:DL15 000(TaKaRa);Lane 1~3:phUb-mRFP cut by NotI,MluIorBamHI,respectively.Fig.1 Lentiviral vector of phUb-mRFP identified by enzymatic digestion
2.2 攜帶mRFP基因慢病毒的包裝、濃縮及成功生產鑒定
將phUb-mRFP與病毒包裝質粒共轉染293FT細胞,48h后倒置熒光顯微鏡下可見紅色熒光,預示轉染成功(圖2A)。收集的病毒上清經超速離心濃縮后感染293FT細胞,48h后倒置熒光顯微鏡下可見紅色熒光(圖2B),預示病毒成功生產;細胞爬片結果顯示濃縮后的病毒可高效率感染293FT細胞(數據未顯示)。以上結果表明mRFP轉基因能夠正常表達(圖2見彩插)。
2.3 mRFP轉基因小鼠的建立
目前,慢病毒載體法制備轉基因小鼠有兩種方法:慢病毒卵周隙注射法和慢病毒感染去透明帶的受精卵法。本研究采用慢病毒卵周隙注射法建立mRFP轉基因小鼠,如圖3所示。
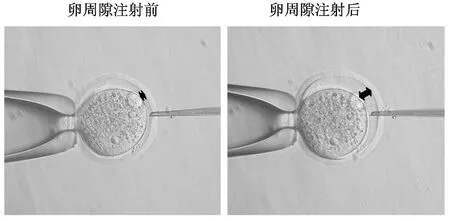
圖3 慢病毒受精卵卵周隙注射示意圖Fig.3 Schematic illustration of subzonal injection(SLI)of lentivirus into a single-cell zygote
本研究先利用體視熒光顯微鏡通過檢測mRFP表達以從出生后的小鼠中篩選與鑒定mRFP轉基因首建鼠,在此基礎上進一步采用PCR進行基因型鑒定以驗證體視熒光顯微鏡鑒定結果。
慢病毒注入48枚單細胞受精卵的卵周隙后,存活胚胎42枚,將其中40枚胚胎移植給2只假孕母鼠,2只均懷孕[受孕率為100%(2/2)],共獲仔鼠6只[產仔率為15%(6/40)]。
將出生幾天的子代乳鼠置于體視熒光顯微鏡下,觀察發現其中兩只強表達mRFP(圖4)。小鼠出生3周后,利用體視熒光顯微鏡檢測mRFP表達,在蛋白水平證實6只出生的F0代中,2只(R3和R4)鼠耳強表達mRFP,其余的弱表達mRFP(R1,R2和R5 R6)或熒光強度(R6)與野生型ICR小鼠無明顯差別(圖5A)。
DNA水平檢測證實,6只F0代中,5只(其中包括強表達mRFP的2只)基因組中整合有外源基因mRFP,即獲得5只PCR陽性的mRFP轉基因首建鼠,首建鼠mRFP整合率達83%(圖5B)。可見,采用PCR進行基因型鑒定很好驗證了體視熒光顯微鏡鑒定結果。
2.4 mRFP轉基因遺傳和表達穩定性檢測
為證實首建鼠所攜帶的外源基因可否穩定遺傳,將mRFP表達陽性及PCR陽性的首建鼠(R3和R4)與野生型ICR鼠交配以獲得F1代。小動物活體成像儀檢測顯示,F1代鼠中部分個體表達mRFP(圖6),這預示外源基因不僅可以從一代向下一代穩定傳遞,且能夠穩定表達(圖2、4~6見彩插1)。
3 討論
3.1 探索非損傷可視轉基因跟蹤策略與方法
轉基因小鼠制作以及隨后的培育、純合子獲得和建系等都是費事、費時和費力的工作。如何將繁重的工作降下來一直是許多研究者探索的問題,我們也在積極探索簡便易行的“非損傷可視外源轉基因跟蹤”策略與方法,以便替代PCR和Southern Blot等傳統跟蹤轉基因的方法。如mRFP轉基因小鼠篩選與鑒定過程中,首先應用體視熒光顯微鏡檢測各組織(鼠耳、皮膚和毛發等)中mRFP表達情況以初步篩選并鑒定mRFP轉基因小鼠,對mRFP表達水平較高的鼠進行基因型鑒定(利用PCR技術),結果顯示這些鼠基因組中確實整合了外源轉基因mRFP。至此,我們初步建立了簡便易行的“非損傷可視外源轉基因跟蹤”策略與方法,該方法有望替代PCR和Southern Blot等傳統跟蹤轉基因的方法。
3.2 慢病毒感染受精卵方式的選擇
慢病毒感染去透明帶的受精卵法的優點:(1)較好控制外源轉基因整合拷貝數;(2)無需特殊儀器和無需熟練的顯微操作技術;(3)適合慢病毒大批量感染受精卵。慢病毒感染去透明帶的受精卵法的缺點:(1)去透明帶時間不易控制,時間稍長即會對胞膜造成損傷;(2)胚胎存活率下降;(3)移植率和妊娠率下降去透明帶法中,胚胎需在體外培養2~3d,在體外過長時間操作,致使胚胎發育延遲,且胚胎在去除透明帶后變得容易相互黏附,移植時胚胎形成聚合體,導致移植率和妊娠率下降。慢病毒卵周隙注射法的優點:(1)胚胎存活率高(因卵周隙注射對細胞膜和核膜均無損傷);(2)移植率和妊娠率高(胚胎有透明帶保護,同時慢病毒注射后胚胎當日或次日即植入假母體內)。慢病毒卵周隙注射法的缺點:(1)外源轉基因整合拷貝數不易控制(因無法精確控制注射入透明帶下慢病毒的量,造成不同首建鼠間外源基因整合拷貝數不一致);(2)需要特殊儀器和熟練操作技術人員。可見,慢病毒受精卵卵周隙注射法和慢病毒感染去透明帶的受精卵法各有優勢,但前者最為常用。
本課題曾嘗試用慢病毒感染去透明帶的受精卵來制備轉基因小鼠,但由于無法克服該方法固有的缺陷,如去透明帶后的胚胎往往粘附聚團,極大延遲胚胎發育,甚至導致胚胎發育停止,從而降低移植率和妊娠率。鑒此,本研究采用透明帶下注射慢病毒法制備轉基因小鼠,以確保得到較高質量的胚胎及較高的移植率和妊娠率等。
3.3 影響mRFP轉基因在轉基因小鼠體內表達因素的分析
在建立轉基因小鼠的過程中,同一轉基因在不同首建鼠,外源轉基因表達水平往往不同,位置效應和轉基因整合拷貝數的不一致通常是造成外源轉基因表達水平有差異的主要原因。本研究數據顯示,在得到的5只mRFP轉基因小鼠中,3只小鼠弱表達mRFP或紅色熒光強度與野生型ICR小鼠無明顯差異,2只小鼠(R3和R4)強表達mRFP,造成如此結果的可能原因有:(1)外源轉基因在染色體上插入位點不同,(2)外源轉基因在基因組中整合拷貝數有差異等。接下來,本研究將從5只mRFP表達水平較強的mRFP轉基因首建鼠中挑選R3和R4進行mRFP轉基因鼠的培育、建系和保種等。
總之,本研究采用慢病毒受精卵卵周隙注射法成功建立mRFP轉基因鼠,得以熟練掌握基于慢病毒載體法制備轉基因小鼠的技術,這為基于慢病毒載體法制備轉基因小鼠,以為活體內研究基因功能提供了技術保障。
[1]Sun Y,Chen X,Xiao D.Tetracycline-inducible expression systems:new strategies and practices in the transgenic mouse modeling[J].Acta Biochim Biophys Sin(Shanghai),2007,39(4):235-246.
[2]Lois C,Hong EJ,Pease S,et al.Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors[J].Science,2002,295(5556):868-872.
[3]Singer O,Tiscornia G,Ikawa M,et al.Rapid generation of knockdown transgenic mice by silencing lentiviral vectors[J].Nat Protoc,2006,1(1):286-292.
[4]Dann CT,Garbers DL.Production of knockdown rats by lentiviral transduction of embryos with short hairpin RNA transgenes[J].MethodsMolBiol,2008,450:193-209.
[5]Rubinson DA,Dillon CP,KwiatkowskiAV,et al.A lentivirusbased system to functionally silence genes in primary mammalian cells,stem cells and transgenic mice by RNA interference[J].Nat Genet,2003,33(3):401-406.
[6]Shin KJ,Wall EA,Zavzavadjian JR,et al.A single lentiviral vector platform formicroRNA-based conditional RNA interference and coordinated transgene expression[J].PNAS,2006,103(37):13759-13764.
[7]Stegmeier F,Hu G,Rickles RJ,et al.A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells[J].PNAS,2005,102:13212-13217.
[8]Szulc J,W iznerowiczM,SauvainMO,et al.A versatile tool for conditional gene expression and knockdown[J].Nat Methods.2006,3(2):109-116.
[9]Tiscornia G,Tergaonkar V,Galimi F,et al.CRE recombinase-inducible RNA interference mediated by lentiviral vectors[J].PNAS,2004,101:7347-7351.
[10]Ventura A,Meissner A,Dillon CP,et al.Cre-lox-regulated conditional RNA interference from transgenes[J].PNAS,2004,101:10380-10385.
[11]Cao F,Lin S,Xie X,et al.In vivo visualization of embryonic stem cell survival,proliferation,and migration after cardiac delivery[J].Circulation,2006,113(7):1005-1014.
[12]賈俊雙,孫妍,肖東,等.慢病毒介導的外源基因體外投遞系統的建立[J].熱帶醫學雜志,2008,8(10):1028-1029,1037.
[13]Nagy A,Gertsenstei M,Vintersten K,et al.Manipulating the mouse Embryo:A Laboratory Manual[J].Cold Spring Harbor Laboratory Press,2004,192-203.
[14]XiaoD,Yue Y,DengXY,et al.Rescue of the albino phenotype by introducing a functional tyrosinaseminigene into Kunming albino mice[J].World J Gastroenterol,2007,13(2):244-249.
[15]Xu K,Deng XY,Yue Y,et al.Generation of the regulatory protein rtTA transgenic mice[J].World J Gastroenterol,2005,8(19):2885-2891.
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
東方少年·布老虎畫刊(2023年8期)2023-08-01 15:45:12
科學大眾(2021年6期)2021-07-20 07:42:44
科學(2020年3期)2020-11-26 08:18:30
學苑創造·A版(2020年9期)2020-10-13 09:41:02
娃娃樂園·綜合智能(2019年3期)2019-04-03 09:17:36
中成藥(2018年2期)2018-05-09 07:19:34
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
湖北師范大學學報(自然科學版)(2015年2期)2016-01-10 08:41:55
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
