PCR方法檢測河豚魚的引物篩選及反應體系優化
2010-03-22 03:40:45陳文炳邵碧英江樹勛李壽崧林河通
食品科學 2010年20期
陳文炳,趙 晨,,邵碧英,江樹勛,閆 誠,李壽崧,林河通
(1.福建出入境檢驗檢疫局,福建 福州 350001;2.福建農林大學食品科學學院,福建 福州 350002)
PCR方法檢測河豚魚的引物篩選及反應體系優化
陳文炳1,趙 晨1,2,邵碧英1,江樹勛1,閆 誠1,李壽崧1,林河通2
(1.福建出入境檢驗檢疫局,福建 福州 350001;2.福建農林大學食品科學學院,福建 福州 350002)
根據Genbank公布的河豚魚細胞色素b基因序列,應用軟件Primer Premier 5.00版設計了7對引物,經過PCR篩選,確定可以在所有8個供試河豚魚樣品中檢出目的DNA片段的引物HT-1,用于建立河豚魚的PCR檢測方法。對該PCR方法中6個因素包括退火溫度、Mg2+終濃度、Taq DNA聚合酶用量、dNTPs終濃度、引物終濃度和模板DNA用量進行優化,確定優化的PCR擴增體系:10×PCR緩沖液2μL,MgCl2終濃度1.5mmol/L,Taq DNA聚合酶1.0U,dNTPs終濃度300μmol/L,引物終濃度0.2μmol/L,DNA模板400ng,加純水至總體積20μL。擴增程序定為94℃預變性5min,94℃變性30s,62℃退火30s,72℃延伸30s,40個循環,72℃延伸5min。據此建立河豚魚成分PCR檢測方法,并通過河豚魚與非河豚魚的PCR檢測結果比較,驗證了該方法的河豚魚特異性。研究結果還表明,該方法的檢出限為0.1%,含量為0.1%的河豚魚樣品PCR檢出率至少在97.5%以上。
河豚魚;引物篩選;PCR檢測;優化
我國海域廣闊,河豚魚種類繁多,魚類加工食品中因混有河豚魚,使得顧客誤食而中毒的事件時有發生,例如2007年6月,發生了由于出口美國的鮟鱇魚混有河豚魚,導致顧客誤食而中毒的事件,對我國海產加工食品出口貿易造成一定負面影響。國外食品檢驗機構也有從魚類加工品中檢出河豚魚的例子,例如2007年4月日本食品檢驗部門在我國南方某食品有限公司出口的馬面魚干中檢出河豚魚成分。為了保證水產加工制品的安全,保障國內外消費者的健康安全,急需一種能夠在食品中快速檢測出河豚魚成分的方法。
PCR檢測方法已被廣泛應用于食品與飼料中的動植物成分與轉基因成分的檢測與鑒定[1-4]。本實驗目的在于參照GenBank公布的河豚魚的線粒體細胞色素b基因DNA序列,設計7對引物,通過PCR試驗篩選,PCR反應體系與溫度條件的優化,建立一種能夠快速檢測海產品中河豚魚源性成分的PCR方法,并測試該方法的檢出限(靈敏度)與檢出率,為此方法的實際應用奠定理論基礎。
1 材料與方法
1.1 材料
8種河豚魚供試材料,收集自福建省廈門、莆田等地水產養殖場,11種常見非河豚海洋魚類,作為陰性對照樣品,購自福州當地超市,美國白魚粉是本實驗室承檢留樣(表1)。
1.2 試劑與儀器
CTAB、Triton-X 100、dNTP 上海博亞生物技術有限公司;Taq DNA聚合酶、10×PCR緩沖液、MgCl225mmol/L Promega公司;動物源性植物飼料基因組DNA提取試劑盒(離心柱型)、蛋白酶K、λDNA/EcoRⅠ+ HindⅢ、DNA MarkerⅠ 天根生化科技(北京)有限公司;瓊脂糖 英國進口的Oxoid公司。
Minispin臺式離心機 Eppendorf公司;Ultrospec 1100 pro核酸蛋白分析儀 Amersham公司;T-Gradient梯度PCR儀 Biometra公司;Power Pac 1000電泳儀 BIORAD公司;Gel Logic 100凝膠成像儀 Kodak公司。
1.3 引物
1.3.1 魚源性成分PCR檢測用引物
[4],由上海生工生物工程有限公司合成一對魚源性成分PCR檢測引物(表2)。

表2 魚源性成分PCR檢測用的引物序列與退火溫度Table2 A pair of primers used in this study for amplifying fish component and corresponding annealing temperature
1.3.2 河豚魚PCR引物設計根據Genbank公布的懷氏兔頭鲀(Lagocephalus wheeleri)和克氏兔頭鲀(Lagocephalus gloveri)線粒體細胞色素b基因(AY128531和EU274423.1)堿基序列,使用引物設計軟件Primer Premier 5.00版設計了7對引物,引物長度在20~25個堿基之間,由上海生工生物技術有限公司合成(表3)。

表3 河豚魚源性成分PCR檢測用的引物序列與退火溫度Table3 Pairs of primers used in this study for amplifying puffer fish component and corresponding annealing temperatures
1.4 DNA提取方法

表1 8種供試河豚魚樣品與12個非河豚魚樣品及其來源Table1 Eight samples of puffer fish, 12 samples of non-puffer fish and their geographical origin
本實驗參照文獻[5-6]的CTAB方法提取河豚魚總DNA,步驟為:稱取50mg樣品于1.5mL離心管中,加入500μL 質量濃度為2g/100mL的CTAB Buffer,10μL蛋白酶K。渦旋30s混勻,于65℃消化3h以上(期間每隔30min應充分振蕩一次)。加入500μL氯仿,渦旋30s混勻后,13000r/min離心10min。取上清液于2mL離心管中,加入2倍體積質量濃度為0.5g/mL 的CTAB沉淀緩沖液,充分混勻后,13000r/min離心10min。取沉淀,加入500μL 1.0mol/L NaCl溶解沉淀。沉淀完全溶解后,加入500μL氯仿。渦旋30s混勻,13000r/min離心10min。取上清液,加入0.8倍體積的異丙醇。渦旋30s混勻,13000r/min離心10min。取沉淀,加入500μL體積分數70%的乙醇,渦旋30s,13000r/min離心10min。取沉淀,用DNA濃縮儀干燥。加入100μL TE(pH 8.0)溶解,備用。
1.5 PCR產物電泳
PCR產物各取10μL,于質量濃度1.5g/100mL的瓊脂糖凝膠電泳。電壓98V,電泳60min,用溴化乙錠染色15min,最后在凝膠成像系統上拍照。
1.6 DNA質量濃度與質量分析方法
1.6.1 DNA質量濃度和純度的測試
用去離子水將DNA稀釋100倍,在核酸蛋白質分析儀上測定其濃度和OD260/OD280值。
1.6.2 PCR擴增效果測試
將供試樣品的DNA稀釋至100ng/μL,用表2魚源性成分PCR引物進行PCR擴增。擴增反應體系為:10× PCR緩沖液2.5μL,25mmol/L MgCl22.5μL,Taq酶1U,10mmol/L dNTPs 0.5μL,模板DNA 1μL,引物終濃度為0.1μmol/L,加純水至25μL。溫度程序為:94℃預變性3min,94℃變性20s,55℃退火40s,72℃延伸40s,40個循環,72℃延伸3min。PCR產物電泳方法按照1.5。
1.7 用于引物篩選的PCR反應體系和溫度循環程序
參考文獻[7-8],將引物篩選的PCR擴增體系初步確定為:10×PCR緩沖液2μL,MgCl2終濃度1.5mmol/L,Taq DNA聚合酶1U,dNTPs終濃度200μmol/L,引物終濃度0.2μmol/L,DNA模板100ng,加純水至20μL。溫度程序定為:94℃預變性5min,94℃變性30s,60℃退火30s,72℃延伸30s,40個循環,72℃延伸5min,4℃保存。按照這個擴增體系與溫度循環程序,用表3的7對引物對8種河豚樣品進行PCR擴增篩選。
1.8 PCR反應體系的優化
以1.7節初步確定的反應體系與溫度循環程序為基礎,對影響PCR擴增的6個因素包括退火溫度、Mg2+濃度、Taq DNA聚合酶用量、dNTPs濃度、引物濃度和模板DNA濃度等進行逐個優化。各因素設置梯度見表4。

表4 影響PCR擴增的6個因素梯度Table4 Gradients of six factors affecting PCR amplification
2 結果與分析
2.1 供試樣品的DNA質量控制
應用CTAB法提取所有供試樣品的DNA,并對其濃度和OD260/OD280值進行測試。20個供試樣品的DNA質量濃度均在571~1007ng/μL范圍內,OD260/OD280值均在1.7~1.9之間,接近純DNA的1.8。魚成分PCR測試結果表明,所有樣品的DNA均可用于PCR檢測(圖1)。
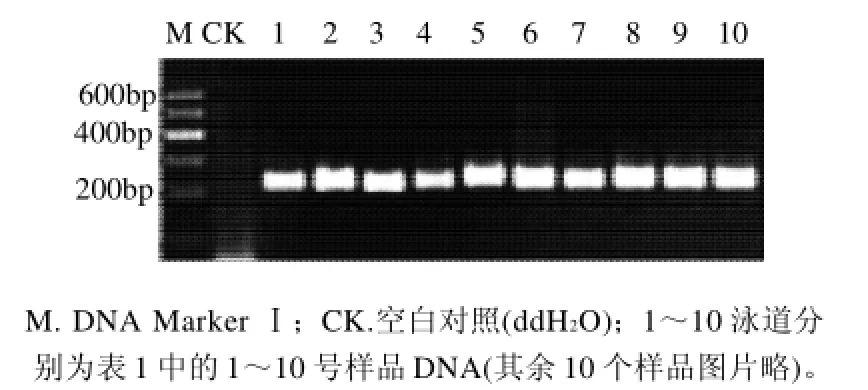
圖1 供試樣品DNA的魚成分PCR擴增結果Fig.1 PCR results of fish component amplified from genomic DNA of 10 test samples
2.2 引物篩選
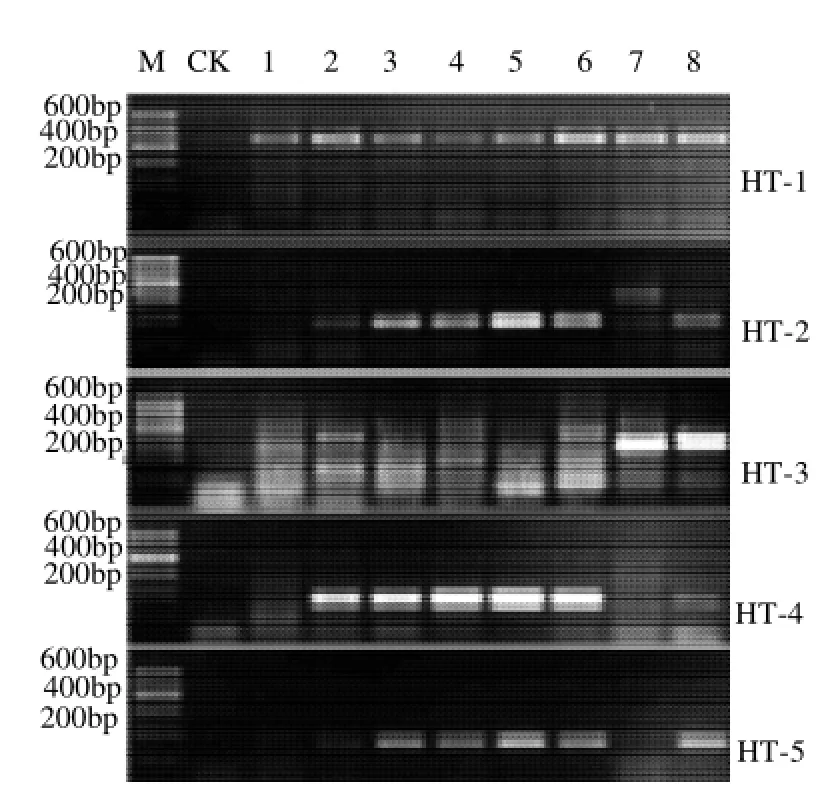
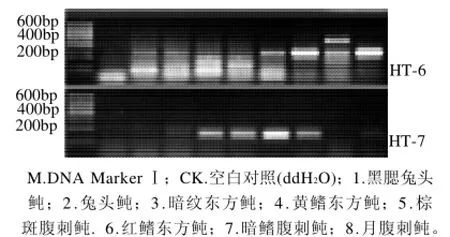
圖2 7對引物(HT-1~HT-7)分別對8個河豚魚樣品的PCR擴增結果Fig.2 PCR results of 8 samples of puffer fishes with 7 pairs of primer HT-1 through HT-7
按照1.7節的PCR反應體系和溫度程序,用表3中的7對引物(HT-1~HT-7)分別對8種河豚樣品進行PCR擴增(圖2)。
由圖2可知,只有引物HT-1能夠擴增出所有供試河豚魚樣品共有的細胞色素b的基因片段,其余引物(HT-2~HT-7)均無法擴增出供試河豚魚樣品共有的DNA片段,因此選取引物HT-1用于后續的PCR檢測方法研究。
2.3 PCR退火溫度及反應體系的優化
2.3.1 退火溫度的優化
對1.7節初步確定的溫度程序中的退火溫度進行優化,設置10個溫度梯度(表4)。PCR擴增結果見圖3。
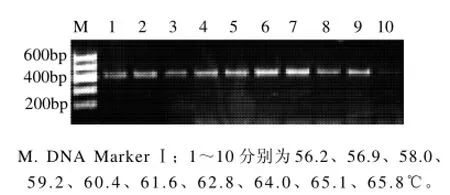
圖3 退火溫度對PCR擴增的影響Fig.3 Effect of annealing temperature on PCR amplification
由圖3可知,除10號泳道的65.8℃ PCR產物條帶亮度明顯較暗外,1~9號的條帶總體上沒有明顯的差異,僅6號泳道的61.6℃與7號的62.8℃的擴增效果相對較佳,因此確定優化的退火溫度為62℃。
2.3.2 Mg2+濃度的優化
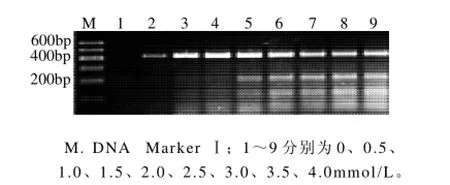
圖4 Mg2+濃度對PCR擴增的影響Fig.4 Effect of Mg2+concentration on PCR amplification
設置9個Mg2+濃度梯度,應用2.3.1節中優化的退火溫度進行擴增,結果見圖4。
由圖4可知,預期的DNA條帶的亮度伴隨Mg2+濃度的變化而變化。Mg2+濃度從0mmol/L到1.5mmol/L,DNA條帶逐漸變亮。Mg2+濃度從2.0mmol/L開始目的DNA片段條帶逐漸變淡,且各泳道在100~300bp之間的位置都出現了2條非預期的DNA雜帶。因此,第4號泳道1.5mmol/L為最佳的Mg2+濃度。
2.3.3 Taq DNA聚合酶用量的優化
按照優化的Mg2+濃度(其他因素按照1.7節方法),配制PCR混合液,以優化后的退火溫度,對Taq DNA聚合酶用量進行優化擴增,結果見圖5。
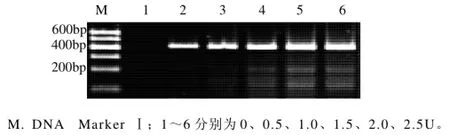
圖5 Taq DNA聚合酶用量對PCR擴增的影響Fig.5 Effect of Taq DNA polymerase amount on PCR amplification
由圖5可知,目的DNA片段條帶的亮度伴隨Taq DNA聚合酶用量的增加而逐漸增強。Taq DNA聚合酶在0U時,沒有任何條帶,在0.5U時出現了清晰的條帶。Taq DNA聚合酶用量為1.5~2.5U之間時,在100~300bp之間的位置都出現了2條預期外的雜帶,且雜帶亮度隨Taq DNA聚合酶用量的增加而增強。顯然由于Taq DNA聚合酶用量過高,導致DNA復制時出現非特異性擴增。為了保證目的條帶的亮度,又能避免預期外雜帶的出現,選用1.0U作為優化的Taq DNA聚合酶用量。
2.3.4 dNTPs濃度的優化
按照已優化的Mg2+濃度和Taq DNA聚合酶用量(其他因素按1.7節),配制PCR混合液,以優化后的退火溫度,對dNTPs濃度進行優化實驗,結果見圖6。

圖6 dNTPs濃度對PCR擴增效果的影響Fig.6 Effect of dNTPs concentration on PCR amplification
由圖6可知,目的DNA條帶的亮度伴隨dNTPs濃度的增加而增強。dNTPs濃度在0μmol/L時,沒有任何條帶,在100μmol/L時就出現了清晰的條帶。當dNTPs濃度達到400μmol/L以上時,在100~300bp之間的位置出現了2條微弱的非預期雜帶。為了保證目的條帶足夠的清晰明亮,且避免非特異擴增雜帶的出現,選用300μmol/L作為優化的dNTPs濃度。
2.3.5 引物濃度的優化
按照已優化的Mg2+濃度、Taq DNA聚合酶用量、dNTPs濃度(其他因素按照1.7節),配制PCR混合液,以優化的退火溫度,對引物濃度進行優化實驗,結果見圖7。
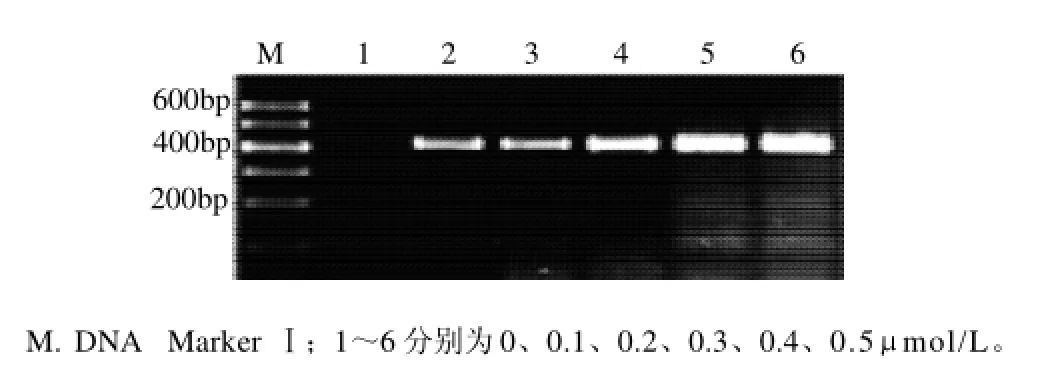
圖7 引物濃度對PCR擴增的影響Fig.7 Effect of primer concentration on PCR amplification
由圖7可知,除了引物濃度在0μmol/L時沒有條帶以外,引物濃度在0.1μmol/L以上的均能在預期位置擴增出明亮、清晰的條帶,且條帶無明顯變化。因此從0.1μmol/L到0.5μmol/L均可滿足PCR對引物濃度的要求。為了保證條帶清晰明亮并考慮到成本的因素,選用0.2μmol/L為優化的引物濃度。
2.3.6 模板DNA用量對擴增的優化
按照優化的Mg2+濃度、TaqDNA聚合酶用量、dNTPs濃度和引物濃度配制PCR混合液,以優化的退火溫度,對模板DNA用量進行優化實驗,結果見圖8。
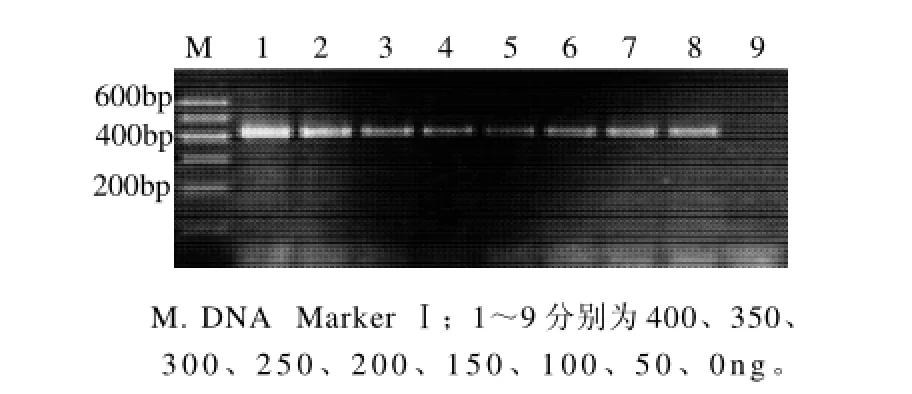
圖8 DNA模板濃度對PCR擴增的影響Fig.8 Effects of DNA template concentration on PCR amplification
由圖8可知,預期的DNA條帶的亮度伴隨模板DNA用量的減少而逐漸變淡。模板DNA用量在400ng時,條帶最亮。模板DNA用量0ng時,沒有任何條帶出現。選用400ng為優化的模板DNA用量。
2.4 優化后的PCR體系與溫度程序的建立以及優化前后的擴增效果比較
2.4.1 優化后的PCR體系與溫度程序的建立
通過對PCR體系和退火溫度的優化,確立最佳的PCR體系和溫度程序:10×PCR緩沖液2μL,MgCl2終濃度1.5mmol/L,Taq DNA聚合酶1.0U,dNTPs終濃度300μmol/L,引物終濃度0.2μmol/L,DNA模板400ng,加純水至總體積20μL。擴增程序定為94℃預變性5min,94℃變性30s,62℃退火30s,72℃延伸30s,40個循環,72℃延伸5min,4℃保存。
2.4.2 優化前后的擴增效果比較
以優化后的PCR體系與溫度程序對供試8種河豚魚樣品DNA進行PCR擴增,同樣取10μL PCR產物進行電泳,獲得的結果(圖9B)比優化前的擴增結果(圖9A)好得多。
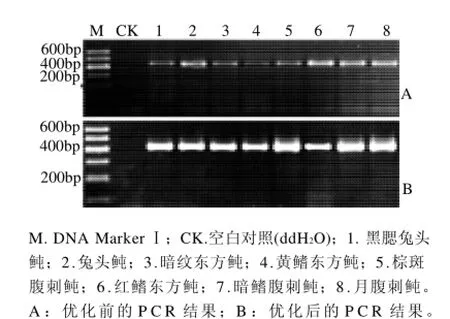
圖9 PCR體系優化前、后的8種河豚魚的PCR結果比較Fig.9 Comparison between PCR results of 8 puffer fish obtained with pre-optimization PCR system and with post-optimization PCR system
2.5 非河豚魚樣品PCR擴增結果
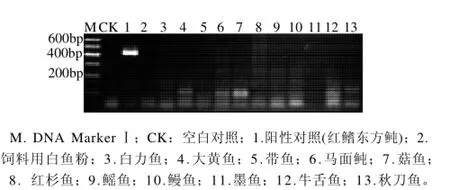
圖10 非河豚樣品的河豚魚成分PCR擴增結果Fig.10 PCR results of puffer fish component in non-puffer fish samples
圖10 顯示,除作為陽性對照樣品(1號泳道)的東方紅鰭鲀(T. rubripes)外,CK與2~13號泳道均未出現目的條帶。這表明,應用引物HT-1能在供試河豚樣品中擴增出預期的DNA條帶,而在空白對照與供試的非河豚樣品中均未能擴增出相應的DNA條帶。可見,引物HT-1對河豚魚有良好的特異性,能夠達到鑒別、區分河豚和非河豚樣品的目的。
2.6 河豚魚成分的檢出限與檢出率
2.6.1 檢出限

圖11 不同河豚魚含量的混合樣品PCR檢測結果Fig.11 PCR results of mixed samples with different contents of puffer fish
從圖11可以看出,含量為100%到0.1%的棕斑腹刺鲀(G. spadiceus)混合樣(以1g河豚粉末與999g陰性美國白魚粉混合)均能擴增出清晰明亮的目的條帶。說明該方法至少可以檢出河豚魚(棕斑腹刺鲀)含量為0.1%的混合樣品中的河豚魚成分。說明該PCR方法的河豚魚成分檢測限(靈敏度)為0.1%。
2.6.2 檢出率
在8個河豚樣品中選取3個樣品,與美國白魚粉混合成河豚魚含量為1%和0.1%的混合樣品,進行檢出率測試,結果見表5。
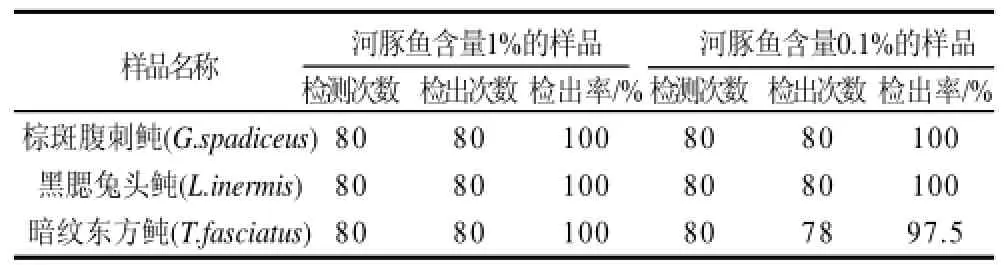
表5 3種河豚不同含量樣品的檢出率Table5 Detection rates in 3 samples with different contents of puffer fish
從表5可知,對于3個河豚魚樣品,該方法在含量為1%時檢出率均能達到達到100%。當河豚含量下降到0.1%時,棕斑腹刺鲀和黑腮兔頭鲀的檢出率仍為100%,暗紋東方鲀檢出率為97.5%,說明該PCR方法有足夠穩定性。
上述PCR分析結果表明,該方法的靈敏度或檢出限為0.1%,3個含量為0.1%的河豚魚樣品經過80次PCR測試,結果表明檢出率都在97.5%以上。
3 討 論
本實驗根據Genbank公布的兩種河豚,即懷氏兔頭鲀(Lagocephalus wheeleri)和克氏兔頭鲀(Lagocephalus gloveri)的線粒體細胞色素b基因DNA序列,設計了7條引物。參考有關文獻[7-8],初步設定PCR反應體系與溫度循環程序,對7對引物進行篩選,發現引物HT-1在供試的8個樣品中都能擴增出目的DNA片段。在該初步設定的PCR反應體系與溫度循環程序基礎上,對影響PCR反應的6個主要因素包括退火溫度、Mg2+濃度、Taq DNA聚合酶用量、dNTPs濃度、引物濃度和模板DNA濃度進行逐一優化。確定優化后的PCR擴增體系:10×PCR緩沖液2μL,MgCl2終濃度1.5mmol/L,Taq DNA聚合酶1.0U,dNTPs終濃度300μmol/L,引物終濃度0.2μmol/L,DNA模板400ng,總體積為20μL。溫度程序定為94℃預變性5min,94℃變性30s,62℃退火30s,72℃延伸30s,40個循環,72℃延伸5min。在PCR反應體系(總體積20μL)與溫度循環程序的優化中,發現Mg2+終濃度超過2.0mmol/L、dNTPs終濃度超過400μmol/L時,都會導致非特異性擴增,出現雜帶,與文獻[9-10]的結果一致。可能是由于Mg2+濃度過高,導致DNA復制時出現非特異性擴增。dNTPs是作為PCR反應的原料參與新鏈DNA的合成,過高濃度的dNTPs會使核苷酸錯誤摻入[11]。本研究還發現Taq DNA聚合酶用量太多(超過1.5U)時,因酶用量過高,導致擴增特異性降低,出現雜帶,與文獻[12]的結果一致。
通過優化前、后PCR擴增效果的對比,發現優化后的擴增效果明顯比優化前的好。以優化后的PCR體系與溫度程序對河豚魚陽性樣品與非河豚魚陰性樣品進行PCR檢測,獲得預期的理想結果,表明本實驗建立的PCR方法對供試的河豚魚樣品具有特異性。根據棕斑腹刺鲀河豚魚樣品不同含量梯度的PCR分析表明,該方法的靈敏度或檢出限為小于等于0.1%,3個含量為0.1%的河豚魚樣品經過80次PCR測試,結果表明2個樣品檢出率為100%,1個檢出率為97.5%,這些結果說明了該PCR方法的穩定性。可見只要設置一定的重復次數,該方法可應用于食品中河豚魚成分的檢驗檢疫實踐。這些結果為該方法應用于食品中河豚魚成分的檢測實踐提供了重要的科學依據。
參考文獻:
[1]陳文炳, 邵碧英, 廖憲彪, 等. 加工食品中若干動物成分的PCR檢測技術應用研究[J]. 食品科學, 2005, 26(8): 338-342.
[2]陳文炳, 江樹勛, 邵碧英, 等. 常見食用菌中轉基因成分定性PCR檢測方法的建立[J]. 食品科學, 2004, 25(10): 206-210.
[3]陳文炳, 邵碧英, 江樹勛, 等. 食品中若干植物源性成分的PCR檢測[J]. 食品科學, 2006, 27(11): 404-408.
[4]張舒亞, 管薇薇, 劉月明, 等. 飼料中魚源性成分真實性鑒別研究[J].飼料研究, 2009(6): 45-46; 48.
[5]MADE D, DEGNER C, GROHMANN L. Detection of genetically modified rice: a construct-specific real-time PCR method based on DNA sequences from transgenic Bt rice[J]. European Food Research and Technology, 2006, 224(2): 271-278.
[6]ALJANABI S M, MARTINEZ I. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques[J]. Nucleic Acids Res, 1997, 25(22): 4692-4693.
[7]劉昊, 連春, 王長康, 等. 豬源性成分的PCR檢測技術優化研究[J].中國農學通報, 2009, 25(18): 1-6.
[8]KYLE C J, WILSON C C. Mitochondrial DNA identification of game and harvested freshwater fish species[J]. Forensic Science International, 2007, 166: 68-76.
[9]馬玉堃, 國會艷, 牛黎明. 雀形目38種鳥類線粒體cytb的引物設計及PCR優化[J]. 林業科技, 2005, 30(6): 29-32.
[10]張寧寧, 劉全蘭, 李磊. 小麥族擬鵝觀草屬Adh基因PCR擴增條件優化[J]. 科學技術與工程, 2009, 9(6): 1397-1401.
[11]田明禮, 吳孝兵, 揚子鱷. SRAP-PCR反應體系的優化及引物篩選[J]. 安徽師范大學學報: 自然科學版, 2008, 31(2): 163-167.
[12]劉志國, 屈伸. 基因克隆的分子基礎與工程原理[M]. 北京: 化學工業出版社, 2003: 75-78.
Primer Screening and Optimization of PCR System for the Detection of Puffer Fish
CHEN Wen-bing1,ZHAO Chen1,2,SHAO Bi-ying1,JIANG Shu-xun1,YAN Cheng1,LI Shou-song1,LIN He-tong2
(1. Fujian Entry-exit Inspection and Quarantine Bureau, Fuzhou 350001, China;2. College of Food Science, Fujian Agriculture and Forestry University, Fuzhou 350002, China)
According to the sequence of cytochrome b gene of puffer fish published in GenBank, seven pairs of puffer fishspecific primers were designed with Primer Premier 5.00 version. After PCR screening, the primer HT-1, which could detect the target DNA fragment in eight samples of puffer fish from different aquatic breeding farm in Fujian province, was selected to establish a PCR method for the detection of puffer fish in this work. Furthermore, six key factors affecting PCR including Taq enzyme and template DNA amounts as well as final magnesiumion (Mg2+), dNTPs and primer concentrations were optimized in order to establish optimal PCR system. And the optimal composition of a 20μL PCR system in water consisted of 2μL of 10 × PCR buffer, 1.5 mmol/L MgCl2, 1.0 U Taq DNA polymerase, 300μmol/L dNTPs, 0.2μmol/L primer and 400 ng of template DNA. The thermal program for PCR was as follows: predenaturation at 94 ℃ for 5 min, denaturation at 94 ℃ for 30 sec, annealing at 62 ℃ for 30 s, polymerization at 72 ℃ for 30 s, and after 40 cycles, final polymerization at 72 ℃ for 5 min. The proposed PCR method was found to be specific through the comparison of PCR results amplified from puffer fish DNA and non-puffer fish DNA. The detection limit was 0.1%, and PCR detection rate for 0.1% content of puffer fish samples was 97.5% or more.
puffer fish;primer screening;PCR detection;optimization
S917.4;Q78
A
1002-6630(2010)20-0376-06
2010-06-08
國家質量監督檢驗檢疫總局科技項目(2008IK175)
陳文炳(1962—),男,研究員,博士,主要從事農產品、食品分子生物學檢測技術研究。E-mail:621213wbc@163.com
猜你喜歡
房地產導刊(2022年5期)2022-06-01 06:20:14
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
建材發展導向(2021年12期)2021-07-22 08:06:48
建材發展導向(2021年7期)2021-07-16 07:07:52
中學生數理化(高中版.高二數學)(2021年12期)2021-04-26 07:43:48
中學生數理化(高中版.高考數學)(2021年12期)2021-03-08 01:28:50
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
