
柴胡炮制后皂苷成分的變化分析
2010-05-26 06:14:40孫秋實(shí)
中成藥 2010年5期
陳 帥, 李 燕, 孫秋實(shí), 楊 方, 李 悅, 吳 彤
(上海醫(yī)藥工業(yè)研究院,上海 200040)
柴胡為傘形科植物柴胡Bupleurum chinenseDC.或狹葉柴胡Bupleurum scorzonerifoliumWild.的干燥根,味苦,微寒,歸肝、膽經(jīng),具有疏散退熱,舒肝、升陽(yáng)的作用[1]。歷代本草和現(xiàn)代中醫(yī)的肝病用藥中多以柴胡為君藥。但柴胡的不同炮制品治療病癥側(cè)重點(diǎn)不一,在用于治療肝病時(shí),常用其醋制品,2005版《中國(guó)藥典》中就收載了醋炙柴胡這一炮制品種。對(duì)于中藥炮制,國(guó)內(nèi)研究主要集中在炮制原理的探究和炮制方法的改進(jìn)等方面[2],對(duì)炮制前后的藥材化學(xué)成分變化的研究較少,僅局限于針對(duì)某一種成分的變化進(jìn)行研究,如總皂苷、揮發(fā)油[3]等。本實(shí)驗(yàn)對(duì)柴胡炮制后及皂化反應(yīng)后皂苷成分的變化進(jìn)行了系統(tǒng)的分析研究,結(jié)果顯示炮制后,柴胡中總皂苷的含量都有所下降,柴胡皂苷b2的含量都大幅增加,柴胡皂苷a、c、d以及a+c+d的含量略微有所減少。皂化反應(yīng)后,柴胡皂苷a+c+d的含量變化很小,柴胡皂苷b2的含量都大幅增加。揭示了柴胡炮制后皂苷類化合物變化規(guī)律,為柴胡飲片的炮制及質(zhì)控提供較為全面的參考。
1 儀器、材料和試劑
1.1 儀器、材料 UV-8500紫外分光光度儀;HP1100高效液相色譜系統(tǒng):G1322A脫氣機(jī),G1316A柱溫箱,G1311A四元泵,G1314A VWD紫外檢測(cè)器。
1.2 試劑 甲醇、乙腈、乙醇、氫氧化鈉、氫氧化鉀、磷酸、碳酸鈉、對(duì)二甲氨基苯甲醛、氨水、正丁醇、磷酸二氫鉀均為分析純;水(超純水)。柴胡皂苷a、c、d、b2均為本實(shí)驗(yàn)室自制(純度大于98%)。
1.3 藥材 見表1。

表1 柴胡及醋制柴胡藥材編號(hào)及產(chǎn)地Tab.1 Serial numbers and places of production of the medicinal materials and the processed bupleurum
2 方法與結(jié)果
2.1 柴胡皂苷b2的含量測(cè)定
2.1.1 分析條件
色譜柱:Diamonsil C18(250 mm×4.6 mm);流速:1 mL/min;柱溫:35℃;檢測(cè)波長(zhǎng):248 nm。見表2。

表2 乙腈-水梯度洗脫條件Tab.2 Gradient elution program of acetonitrile and water
2.1.2 提取方法的考察
2.1.2.1 提取方式的選擇
取CH-5號(hào)藥材0.1 g兩份,精密稱量,置于茄形瓶中,一份精密的加入20 mL 5%氨水甲醇溶液于80℃水浴回流2 h,過濾,蒸干,精密加入5 mL甲醇溶液定容;另一份于試管中超聲兩次,每次加入5%氨水甲醇溶液10 mL,超聲1 h,離心,將兩次上清液合并蒸干,精密加入5 mL甲醇溶液定容,按2.1.1項(xiàng)色譜條件進(jìn)行分析,實(shí)驗(yàn)結(jié)果顯示柴胡皂苷b2回流的提取效果優(yōu)于超聲,所以選擇回流這種提取方式。
2.1.2.2 提取時(shí)間的選擇
取CH-5號(hào)藥材0.1 g,精密稱量,置茄形瓶中,分別精密的加入20 mL 5%氨水甲醇溶液于80℃水浴回流,分別1 h、2 h、3 h、4 h,回流后過濾,蒸干,分別精密加入5 mL甲醇溶液定容,進(jìn)樣,按2.1.1項(xiàng)色譜條件進(jìn)行HPLC分析。1 h、2 h、3 h、4 h提取后含量幾乎沒有明顯的變化,為了既節(jié)約時(shí)間又使其充分的回流,并結(jié)合文獻(xiàn)的報(bào)道,選擇2 h為回流提取的時(shí)間。
2.1.2.3 提取次數(shù)的選擇
取CH-5號(hào)藥材0.1 g兩份,精密稱量,置于茄形瓶中,分別精密的加入5%氨水甲醇溶液20 mL于80℃水浴回流,一份回流2 h,過濾,蒸干,精密加入5 mL甲醇溶液,定容;另一份先回流2 h后,將上清液過濾,再加入20 mL 5%氨水甲醇溶液于濾渣中,再回流2 h,合并兩次濾液,蒸干,精密加入5 mL甲醇溶液,定容,按2.1.1項(xiàng)色譜條件進(jìn)行HPLC分析。結(jié)果顯示回流一次和兩次對(duì)于柴胡皂苷b2的提取效果并無明顯差異,所以選擇回流1次。
2.1.2.4 提取溶劑的選擇
柴胡提取溶劑的選擇取CH-5號(hào)藥材0.1 g兩份,精密稱量,置于茄形瓶中,一份加入5%氨水甲醇溶液20 mL,另一份加入5%氨水95%乙醇溶液20 mL,于80℃水浴回流2 h,過濾,蒸干,精密加入5 mL甲醇溶液,定容,按2.1.1項(xiàng)色譜條件進(jìn)行分析。結(jié)果顯示對(duì)于柴胡皂苷b2的提取,加入5%氨水甲醇溶液的提取效果優(yōu)于加入5%氨水95%乙醇溶液,所以對(duì)于柴胡的提取選擇加入5%氨水甲醇溶液。
醋制柴胡提取溶劑的選擇 取CH-5-4號(hào)藥材0.1 g 4份,精密稱量,置于茄形瓶中,分別精密的加入20 mL甲醇、5%氨水甲醇、95%乙醇、5%氨水95%乙醇溶液于80℃水浴回流2 h,過濾,蒸干,精密加入5 mL甲醇溶液定容,按2.1.1項(xiàng)色譜條件進(jìn)行分析,結(jié)果顯示5%氨水甲醇溶液對(duì)醋柴胡中柴胡皂苷b2的提取效果也較好。
綜上,得出柴胡皂苷b2的最佳提取方法:藥材粉末0.1 g,加入5%氨水甲醇溶液20 mL,于水浴回流2 h。
2.1.3 含量測(cè)定
取不同柴胡樣品,按供試品溶液制備方法操作并按2.1.1項(xiàng)色譜條件進(jìn)行HPLC分析,記錄柴胡皂苷b2的峰面積,計(jì)算含量,結(jié)果見表3。見圖1~4。
柴胡皂苷b2的含量測(cè)定結(jié)果顯示柴胡炮制后,柴胡皂苷b2的含量從10.7%~78.2%都有不同程度的提高。
2.1.4 方法學(xué)考察
2.1.4.1 對(duì)照品溶液的制備

表3 不同批次柴胡中柴胡皂苷b2的含量Tab.3 Content determination of saikosaponin b2of various samples

圖1 對(duì)照品柴胡皂苷b2HPLC圖譜Fig.1 HPLC chromatogram of saikosaponin b2 reference substance

圖2 藥材CH-5中柴胡皂苷b2含量測(cè)定HPLC圖譜Fig.2 HPLC chromatogram of saikosaponin b2in medicinal material CH-5

圖3 藥材CH-5-1中柴胡皂苷b2含量測(cè)定HPLC圖譜Fig.3 HPLC chromatogram of saikosaponin b2in medicinal material CH-5-1
柴胡皂苷b2自制。取柴胡皂苷b2對(duì)照品適量,精密稱量,加甲醇制成每1 mL中含0.067 8 mg的溶液,即得。
2.1.4.2 供試品溶液的制備
取柴胡藥材0.1 g,精密稱量,置于茄形瓶中,分別精密的加入5%氨水甲醇溶液20 mL于80℃水浴回流2 h,將濾液過濾,蒸干,精密加入5 mL甲醇溶液定容,即為供試品溶液。

圖4 藥材CH-5-2中柴胡皂苷b2含量測(cè)定HPLC圖譜Fig.4 HPLC chromatogram of saikosaponin b2in medicinal material CH-5-2
2.1.4.3 標(biāo)準(zhǔn)曲線
取柴胡皂苷b2對(duì)照品溶液,精密吸取1.0、2.5、5.0、10.0、15.0、20.0 μL,按 2.1.1 項(xiàng)色譜條件進(jìn)行HPLC分析,以進(jìn)樣量(μg)為橫坐標(biāo),峰面積值(A)為縱坐標(biāo)繪制標(biāo)準(zhǔn)曲線,得回歸方程:A=75.6C-8.90,R2=1。結(jié)果表明柴胡皂苷b2在0.067 8~1.36 μg范圍內(nèi),進(jìn)樣量與峰面積之間線性關(guān)系良好。
2.1.4.4 精密度
取對(duì)照品溶液,在2.1.1項(xiàng)色譜條件下進(jìn)行測(cè)定,連續(xù)進(jìn)樣5次,記錄柴胡皂苷b2的峰面積,并計(jì)算相對(duì)標(biāo)準(zhǔn)偏差,RSD為0.69%。
2.1.4.5 重復(fù)性
取樣品CH-5-1,5份,每份0.1 g,精密稱定,照供試品溶液制備方法分別制成供試品溶液,按2.1.1項(xiàng)色譜條件測(cè)定,記錄柴胡皂苷b2的峰面積,計(jì)算相對(duì)標(biāo)準(zhǔn)偏差,RSD為0.49%。
2.1.4.6 穩(wěn)定性
取樣品CH-5-1,0.1 g,精密稱定,照供試品溶液制備方法制成供試品溶液,按2.1.1項(xiàng)色譜條件分別于0、2、4、6、8 h 測(cè)定,記錄柴胡皂苷 b2的峰面積,計(jì)算相對(duì)標(biāo)準(zhǔn)偏差,RSD為1.7%,表明樣品在8 h內(nèi)穩(wěn)定。
2.1.4.7 加樣回收率
取6份已知含量的同一批柴胡樣品CH-5-1,各0.05 g,精密稱定,分別加入對(duì)照品溶液0.067 8 mg/mL,3.7、5、6 mL,按供試品溶液制備方法制備樣品試液,按2.1.1項(xiàng)色譜條件進(jìn)樣分析,記錄柴胡皂苷b2的峰面積并計(jì)算其含量,平均加樣回收率為97.4%,RSD為2.6%。
2.2 柴胡皂苷a、c、d的含量測(cè)定
2.2.1 分析條件
色譜柱、流動(dòng)相、流速和柱溫條件參考2.1.1項(xiàng);檢測(cè)波長(zhǎng):204 nm。
2.2.2 對(duì)照品溶液的制備
柴胡皂苷a、c、d均為實(shí)驗(yàn)室自制。分別取柴胡皂苷a、c、d對(duì)照品適量,精密稱定,加甲醇制成濃度分別為 0.216、1.44、0.278 mg/mL的溶液,即得。分別取柴胡皂苷 a、c、d,0.8、0.09、1 mL 于 2 mL 量瓶中,用甲醇定容,得到混合對(duì)照品溶液Ⅰ,取混合對(duì)照品溶液Ⅰ0.5 mL于1 mL量瓶中,用甲醇定容,得到混合對(duì)照品溶液Ⅱ。即每1 mL混合對(duì)照品溶液Ⅱ中分別含柴胡皂苷 a、c、d為 0.008 64 mg、0.006 48 mg、0.013 9 mg。
2.2.3 供試品溶液的制備
取柴胡藥材0.1 g于茄形瓶中,精密稱定,分別加入20 mL 5%氨水-甲醇溶液于80℃水浴回流2 h,將濾液過濾蒸干,精密加入10 mL甲醇溶液定容,即為供試品溶液。
2.2.4 含量測(cè)定
取不同批次的柴胡樣品,按供試品溶液制備方法操作并按2.2.1項(xiàng)色譜條件進(jìn)行HPLC分析,記錄柴胡皂苷a、c、d的峰面積,計(jì)算含量,結(jié)果見表4,圖5~8。

表4 不同批次柴胡中柴胡皂苷a、c、d的含量Tab.4 Content determination of saikosaponin a、c、d of various samples

圖5 對(duì)照品柴胡皂苷c、d、a HPLC圖譜Fig.5 HPLC chromatogram of saikosaponin c、d、a reference substance
結(jié)果顯示柴胡皂苷a+c+d的含量在炮制后含量略微有所減少,減少的程度從-0.375%~25.2%。表明原生皂苷在酸的條件下轉(zhuǎn)化為次生皂苷,次生皂苷b2的含量有所增加,而原生皂苷柴胡皂苷a+c+d的含量有所減少。
2.2.5 方法學(xué)考察
2.2.5.1 標(biāo)準(zhǔn)曲線
取柴胡皂苷混合對(duì)照品溶液Ⅱ,精密吸取2.5、5.0、10.0、15.0、20.0 μL,按 2.2.1 項(xiàng)色譜條件進(jìn)行HPLC分析,以進(jìn)樣量(μL)為橫坐標(biāo),峰面積值(A)為縱坐標(biāo)繪制標(biāo)準(zhǔn)曲線,得回歸方程,結(jié)果見表5,表明柴胡皂苷 a、c、d 分別在0.216 ~1.73 μg、0.162~1.30 μg、0.350 ~2.78 μg 范圍內(nèi),進(jìn)樣量與峰面積之間線性關(guān)系良好,符合外標(biāo)法定量測(cè)定要求。
2.2.5.2 精密度

圖6 藥材CH-5中柴胡皂苷c、d、a含量測(cè)定HPLC圖譜Fig.6 HPLC chromatogram of saikosaponin c、d、a in medicinal material CH-5

圖7 藥材CH-5-1中柴胡皂苷c、d、a含量測(cè)定HPLC圖譜Fig.7 HPLC chromatogram of saikosaponin c、d、a in medicinal material CH-5-1
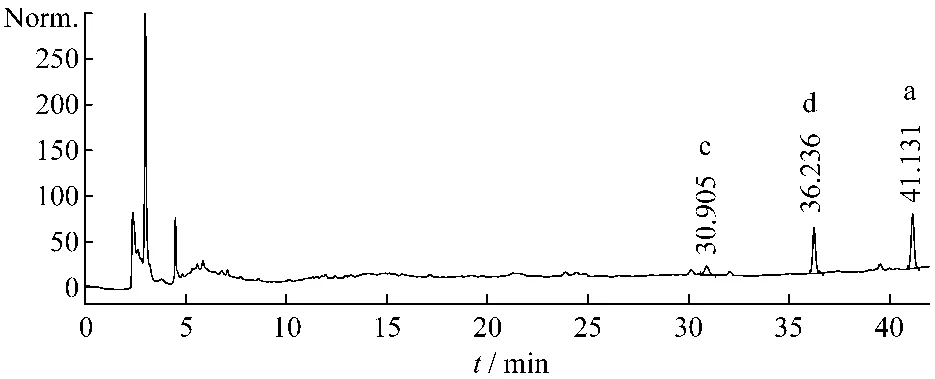
圖8 藥材CH-5-2中柴胡皂苷c、d、a含量測(cè)定HPLC圖譜Fig.8 HPLC chromatogram of saikosaponin c、d、a in medicinal material CH-5-2
表5 柴胡皂苷a,c,d的線性關(guān)系測(cè)定結(jié)果
Tab.5 Results of linear ranges of saikosaponin a、c、d

指標(biāo)成分 回歸方程 R2 線性范圍/μg+3.24 0.999 0.216~1.73柴胡皂苷c Y=20.4X+0.233 0.999 0.162~1.30柴胡皂苷d Y=57.0X柴胡皂苷a Y=46.2X+8.22 0.999 0.350~2.78
取柴胡皂苷混合對(duì)照品溶液Ⅱ,在2.2.1項(xiàng)色譜條件下進(jìn)行HPLC分析,連續(xù)進(jìn)樣5次,記錄柴胡皂苷a、c、d的峰面積并計(jì)算相對(duì)標(biāo)準(zhǔn)偏差,柴胡皂苷 a、c、d 的 RSD 分別為0.36%、0.63%、0.29%,表明色譜系統(tǒng)精密度符合要求。
2.2.5.3 重復(fù)性
取樣品CH-5-2,5份,每份0.1 g,精密稱量,按照供試品溶液制備方法分別制成供試品溶液,按2.2.1項(xiàng)色譜條件測(cè)定,記錄柴胡皂苷a、c、d的峰面積,計(jì)算相對(duì)標(biāo)準(zhǔn)偏差,柴胡皂苷a、c、d的RSD分別為2.0%、1.8%、1.9%,表明方法的重復(fù)性較好。
2.2.5.4 穩(wěn)定性
取樣品CH-5-2,0.1 g,精密稱量,照供試品溶液制備方法分別制成供試品溶液,按2.2.1項(xiàng)色譜條件分別于 0、2、4、6、8 h 測(cè)定,記錄柴胡皂苷 a、c、d的峰面積,計(jì)算相對(duì)標(biāo)準(zhǔn)偏差,柴胡皂苷a、c、d的RSD分別為0.85%、2.1%、1.9%,表明方法的穩(wěn)定性較好。
2.2.5.5 加樣回收率
取5份同一批柴胡樣品 CH-5-2,各0.05 g,精密稱量,分別加入混合對(duì)照品溶液(Ca=0.53 mg/mL,Cc=0.098 mg/mL,Cd=0.60 mg/mL)0.65 mL、0.75 mL、0.85 mL,按供試品溶液制備方法制備樣品試液,按2.2.1項(xiàng)色譜條件進(jìn)行測(cè)定,記錄柴胡皂苷a、c、d的峰面積,計(jì)算相對(duì)標(biāo)準(zhǔn)偏差,柴胡皂苷a、c、d的平均加樣回收率為 94.0%、95.6%、98.9%,RSD分別為2.7%、3.2%、2.3%。
2.3 皂化后皂苷的含量測(cè)定
2.3.1 皂化方法的比較
結(jié)合文獻(xiàn)報(bào)道的兩種皂苷皂化的方法[4,5],采用以下方法進(jìn)行了比較。
2.3.1.1 皂化方法Ⅰ
取柴胡藥材約0.1 g,精密稱量,置于茄形瓶中,加5%氫氧化鉀甲醇20 mL液浸漬過夜,加熱回流5 h,提取液蒸干,加水30 mL,正丁醇溶液萃取(30 mL,30 mL,30 mL),合并萃取液加1%磷酸二氫鉀溶液50 mL洗滌,棄水相,再以40 mL水洗滌1次,有機(jī)相蒸干,精密加入10 mL甲醇定容。用2.1.1項(xiàng)、2.2.1項(xiàng)的分析條件進(jìn)行分析,得到如表6結(jié)果:

表6 皂化反應(yīng)的選擇Tab.6 Selection of saponated action
2.3.1.2 皂化方法Ⅱ
藥材粉末1.0 g,用90%甲醇提取3次,每次15 min,溶劑量分別為 20 mL,15 mL,15 mL,合并 3 次濾液,用90%甲醇定容到50 mL,取其中的5 mL,加入氫氧化鈉溶液2.5 mL于50℃水浴回流1 h,然后加入磷酸二氫鉀-氫氧化鈉緩沖溶液(100 mL 0.2 mol/L磷酸二氫鉀和69.5 mL 0.2 mol/L氫氧化鈉混合)7.5 mL,混合液過減壓ODS柱,先用35%甲醇洗脫10 mL,再用甲醇洗脫,收集甲醇洗脫液10 mL。
藥材粉末0.1 g,用90%甲醇提取3次,每次15 min,溶劑量分別為 2 mL,1.5 mL,1.5 mL,合并3 次濾液,用90%甲醇定容到5 mL,加入氫氧化鈉溶液0.25 mL于50℃水浴回流1 h,然后加入磷酸二氫鉀-氫氧化鈉緩沖溶液(100 mL 0.2 mol/L磷酸二氫鉀和69.5 mL 0.2 mol/L氫氧化鈉混合)0.75 mL,混合液過減壓ODS柱,先用35%甲醇洗脫10 mL,再用甲醇洗脫,收集甲醇洗脫液10 mL。用2.1.1項(xiàng)、2.2.1項(xiàng)的分析條件進(jìn)行分析,得到如表7結(jié)果:

表7 皂化方法Ⅱ的結(jié)果Tab.7 Results of saponated actionⅡ
2.3.1.3 皂化方法Ⅱ的改進(jìn)方法
藥材粉末0.1 g,用90%甲醇提取3次,每次15 min,溶劑量分別為 2 mL,1.5 mL,1.5 mL,合并3 次濾液,用90%甲醇定容到5 mL,加入氫氧化鈉溶液0.25 mL于50℃水浴回流1 h,提取液蒸干,加水30 mL,正丁醇溶液萃取(30 mL,30 mL,30 mL),合并萃取液加1%磷酸二氫鉀溶液50 mL洗滌,棄水相,再以40 mL水洗滌1次,有機(jī)相蒸干,精密加入10 mL甲醇定容。用2.1.1項(xiàng)、2.2.1項(xiàng)的分析條件進(jìn)行分析,得到如表8結(jié)果:
對(duì)比以上3種方法,皂化方法Ⅱ以及皂化方法Ⅱ的改進(jìn)方法都不能將柴胡中的皂苷完全的皂化,而皂化方法Ⅰ所得到的結(jié)果較為理想,所以選擇皂化方法Ⅰ為實(shí)驗(yàn)方法。

表8 皂化方法Ⅱ改進(jìn)方法的結(jié)果Tab.8 Results of improved saponated actionⅡ
2.3.2 供試品溶液的制備
精密稱取柴胡藥材約0.1 g,置于茄形瓶中,加5%氫氧化鉀甲醇20 mL液浸漬過夜,加熱回流5 h,提取液蒸干,加水30 mL,正丁醇溶液萃取(30 mL,30 mL,30 mL),合并萃取液加1%磷酸二氫鉀溶液50 mL洗滌,棄水相,再以40 mL水洗滌1次,有機(jī)相蒸干,加甲醇溶解,定容10 mL,即為供試品溶液。
2.3.3 對(duì)照品溶液的制備
見2.1.4.1項(xiàng),2.2.2項(xiàng)。
2.3.4 含量測(cè)定
將柴胡的各個(gè)批次的藥材制備成供試品,按照2.1.1項(xiàng)、2.2.1項(xiàng)分析條件進(jìn)行分析,結(jié)果見表9。
2.3.5 皂化反應(yīng)前后總皂苷的含量測(cè)定,分析方法參考文獻(xiàn)[6,7],方法學(xué)考察略。
皂化反應(yīng)后,柴胡皂苷a、c、d以及a+c+d的含量在皂化反應(yīng)前后變化幅度不大,但是柴胡皂苷b2的含量卻大幅增加,增加了185%,而總皂苷的含量在皂化后卻有所減少,減少了17.2%;醋炙柴胡、醋拌柴胡,在皂化反應(yīng)后柴胡皂苷a、c、d以及a+c+d的含量變化并不大,大部分都有所減少,柴胡皂苷b2的含量幾乎都有所增加,增加的幅度從24.6%~95.9%不等。總皂苷的含量也是增減不一,說明炮制品總皂苷的含量在皂化反應(yīng)后無明顯變化;對(duì)照藥材,在皂化反應(yīng)后,柴胡皂苷a+c+d的含量略微有所減少,但是柴胡皂苷b2的含量卻大幅的增加,增加了117%,而總皂苷的含量在皂化反應(yīng)后卻有所減少,減少了49.9%;對(duì)照藥材的炮制品醋拌柴胡,柴胡皂苷a、c、d以及a+c+d的含量在皂化反應(yīng)后大幅度的減少,減少了72.2%,柴胡皂苷b2的含量也大幅度減少,減少了71.5%,總皂苷的含量也減少了30.4%。見表10。
3 討論與結(jié)論
本實(shí)驗(yàn)首次采用皂化反應(yīng)測(cè)定柴胡炮制前后皂苷的含量變化,前人研究表明柴胡含有大量的乙酰化皂苷成分[8],僅以柴胡皂苷 a、c、d、b2、b1作為對(duì)照,其結(jié)果有一定偏差,不能完全代表藥材的實(shí)際含量,通過皂化反應(yīng)去乙酰化后,分析結(jié)果較全面。在文獻(xiàn)的基礎(chǔ)上確定柴胡皂化反應(yīng)方法,該法操作簡(jiǎn)單、重現(xiàn)性好、精密度良好,可以用于柴胡及其炮制品的皂苷成分含量分析。
通過結(jié)果說明,炮制后,總皂苷的含量都有所下降,柴胡皂苷b2的含量都大幅增加,柴胡皂苷a、c、d以及a+c+d的含量并沒有什么變化。說明柴胡中的原生皂苷柴胡皂苷a、c、d在植物中的酸性成分條件下轉(zhuǎn)化為次生苷柴胡皂苷b2、b1時(shí),接近一半的柴胡皂苷b2、b1上面的羥基被乙酰化,而皂化反應(yīng)后,乙酰基脫去,又重新轉(zhuǎn)化為柴胡皂苷b2,所以含量大幅增加。而柴胡的炮制品中,可能被乙酰化的柴胡皂苷b2部分被去乙酰化,但并不完全,所以柴胡皂苷b2增加的幅度并沒有柴胡大。以上的研究初步揭示了柴胡炮制后皂苷類化合物變化規(guī)律,為柴胡飲片的炮制及質(zhì)控提供較為全面的參考。
柴胡炮制前后化學(xué)成分的比較分析,顯示醋炙、醋拌這兩種炮制工藝對(duì)柴胡總皂苷的含量變化沒有顯著影響,醋炙、醋拌是否可以通用,還需對(duì)其炮制后化學(xué)成分組成及其量比關(guān)系的變化進(jìn)一步研究。

表9 柴胡各個(gè)樣品皂化后皂苷的含量Tab.9 Results of saponated action

表10 柴胡各個(gè)樣品皂化后總皂苷的含量Tab.10 Content of total saponins after saponated action
[1]中國(guó)藥典[S].一部.2005:198.
[2]葉定江,原思通.中藥炮制學(xué)辭典[M].上海:上海科學(xué)技術(shù)出版社,2005:295-296.
[3]劉 偉,黃國(guó)理.柴胡的不同炮制方法對(duì)其有效成分影響的研究[J].中藥材,2005,18(1):21-23.
[4]李玲玲,張水龍.黃芪中有效成分環(huán)黃芪醇皂甙和黃芪甲甙的含量測(cè)定[J].中國(guó)現(xiàn)代應(yīng)用藥學(xué),1998,15(3):13-15.
[5]Suzuki H,Yokota Y,Terasaki S,et al.Component Determination of Total Saponins in Bupleurum Root by High-performance Liquid Chromatography[J].Nat Med,2004,58(4):138-144.
[6]浦錦寶,胡軼娟,佘 靖,等.紫外可見分光光度法測(cè)定柴胡中柴胡總皂苷的含量[J].醫(yī)學(xué)研究雜志,2008,37(6):98-100.
[7]鄭虎占,董澤宏,佘 靖.中藥現(xiàn)代研究與應(yīng)用(第四卷)[M].北京:學(xué)苑出版社,1998:3688-3689.
[8]劉沁舡,譚 利,白焱晶,等.柴胡屬植物皂苷近10年研究概況[J].中國(guó)中藥雜志,2002,27(1):7-11.
