
高效液相色譜-二極管陣列-離子阱質譜法對紅花水提物中有效成分群的定性分析*
2010-07-29 13:45:54王曉雯張亞軍趙新鋒
中國藥業 2010年15期
王曉雯 ,彭 寧 ,張 鵬 ,左 燕 ,張亞軍 ,趙新鋒
(1.陜西省人民醫院藥劑科,陜西 西安 710068; 2.西北大學生命科學學院,陜西 西安 710069)
紅花為菊科一年生草本植物紅花 Carthamus tinctorius L.的干燥花,具有活血通經、散瘀止痛的功效[1]。藥理研究表明,黃酮類和色素類成分為紅花擴張血管、增加冠脈流量和改善心腦血液循環的主要有效成分[2]。趙明波等[3]以羥基紅花黃色素A為基準物,對不同產地的紅花藥材進行了指紋圖譜分析。但除了基準物,上述研究未能對色譜峰進行歸屬,其原因在于缺少紅花藥材有效成分群的定性分析方法。筆者采用高效液相色譜-二極管陣列-離子阱質譜(HPLC-DAD-MSn)法對紅花水提物中的黃酮類成分進行了定性分析,旨在為紅花指紋圖譜研究中色譜峰的歸屬提供參考,現報道如下。
1 儀器與試藥
Agilent 1100系列高效液相色譜儀(美國安捷倫公司),包括在線真空脫氣機、二元梯度泵、柱溫箱、二極管陣列檢測器;SL型離子阱質譜(美國安捷倫公司);Maxima超純水機(美國基因公司);Sartorius BP221S型萬分之一電子天平(美國Sartorius公司)。紅花藥材(新疆產,經陜西省食品藥品檢驗所鑒定);槲皮素化學對照品(中國藥品生物制品檢定所,批號為100081-200406);羥基紅花黃色素A化學對照品(中國藥品生物制品檢定所,批號為11637-200503);色譜純乙腈(Fisher公司,美國),超純蒸餾水(自制),其他試劑均為分析純。
2 方法與結果
2.1 色譜條件
色譜柱:Agilent SB-C18柱(150 mm ×2.1 mm,5 μm);流速:0.2 mL/min;柱溫:30℃;檢測波長:0~10.0 min為280 nm,10.0~25.0 min為 370 nm,25.0~45.0 min為 430 nm;流動相:A為0.2%的甲酸水溶液,B為甲醇,梯度洗脫,0~20.0 min為30%B,20.0~30.0 min為 30%B→65%B,30.0~45.0 min為 65%B。在此條件下,紅花水提物的高效液相色譜圖見圖1。
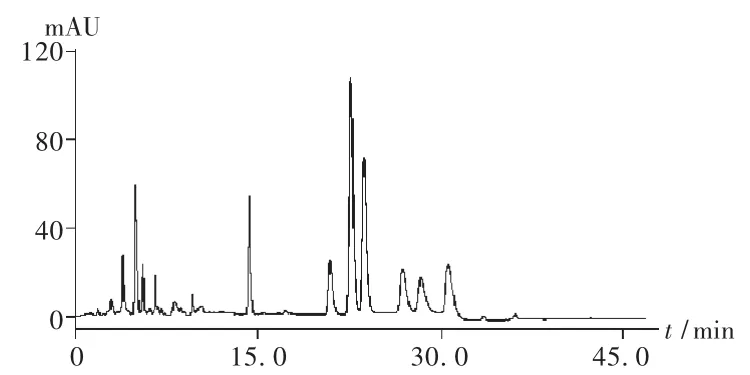
圖1 紅花水提物高效液相色譜圖
2.2 質譜條件
電噴霧負離子模式(ESI-)電離,霧化氣壓強為275 kPa,干燥氣流速為7.0 mL/min,干燥氣溫度為325℃,毛細管電壓為-4200 V,掃描范圍為50~1500 amu。在此條件下,紅花水提物的ESI-MS總離子流圖見圖2。
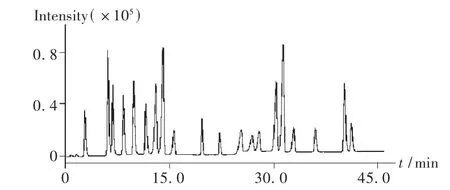
圖2 紅花水提物總離子流圖
2.3 溶液制備
分別精密稱取槲皮素和羥基紅花黃色素A對照品0.012 g和0.016 g,以30.0%的甲醇水溶液溶解,分別置2個25.0 mL具塞量瓶中,加甲醇至刻度,制備成質量濃度分別為0.48 g/L和0.64 g/L的對照品溶液,4℃冷藏備用。取干燥至恒重的紅花藥材10.0 g,置250 mL圓底燒瓶中,加10倍量水,加熱回流1.5 h,重復3次,抽濾,合并濾液,減壓濃縮至膏狀物,真空干燥,研磨成粉,以30.0%的甲醇水溶液定容于500.0 mL具塞量瓶中,即紅花藥材供試品溶液。4℃冷藏備用。
2.4 方法學考察
線性關系考察:分別精密吸取2.3項下槲皮素和羥基紅花黃色素A對照品溶液適量,用30.0%的甲醇水溶液稀釋為系列對照品溶液。在擬訂的分析條件下,分別進樣10.0 μL,對離子阱質譜的多級反應模式(MRM)進行分析。槲皮素選擇 m/z 301.1→m/z 283.0為離子對,羥基紅花黃色素A選擇 m/z 611.2→m/z 449.1為離子對,測定提取離子流圖的峰面積。分別以槲皮素和羥基紅花黃色素A提取離子流圖的峰面積對其質量濃度進行線性回歸,得回歸方程分別為 Y=12.4 X+1.2(r=0.9998)和 Y=5.1 X+0.4(r=0.9997),質量濃度線性范圍分別為0.48~48.0 μg/mL和0.64~320 μg/mL。
精密度試驗:按2.3項下供試品溶液制備方法制備供試品溶液,用30%甲醇水溶液稀釋10倍后,連續進樣5次,每次10.0 μL,在擬訂的分析條件下,分別測定槲皮素和羥基紅花黃色素A的提取離子流圖的峰面積,計算得其標準偏差 RSD分別為1.2%和0.87%。
穩定性試驗:按2.3項下供試品溶液制備方法制備供試品溶液,以30%甲醇水溶液稀釋10倍后,每2 h進樣1次,每次10.0 μL,在擬訂的分析條件下測定峰面積。結果48 h內槲皮素和羥基紅花黃色素A峰面積的 RSD分別為1.3%和1.0%,表明供試品溶液在48 h內具有良好的穩定性。
重復性試驗:按2.3項下供試品溶液制備方法制備5份供試品溶液,以30%甲醇水溶液稀釋10倍后,分別進樣,每份每次10.0 μL,在擬訂的分析條件下測定峰面積。結果槲皮素和羥基紅花黃色素A峰面積的 RSD分別為1.1%和1.2%。
回收率試驗:取適量已知槲皮素和羥基紅花黃色素A含量的紅花藥材5份,精密加入2.3項下槲皮素和羥基紅花黃色素A對照品溶液適量后,按2.3項下方法制備成供試品溶液。以30%甲醇水溶液稀釋10倍后,在擬訂的分析條件下分別進樣10.0 μL,測定峰面積,計算回收率。結果槲皮素和羥基紅花黃色素A的平均回收率分別為98.7%和99.2%。
2.5 樣品含量測定
按2.3項下供試品溶液制備方法制備紅花供試品溶液5份,以30%甲醇水溶液稀釋10倍后,在擬訂的分析條件下進樣,外標法計算含量。結果紅花藥材中槲皮素和羥基紅花黃色素A的平均含量分別為0.13%和1.14%。
2.6 有效成分群定性分析
取2.3項下制備的供試品溶液,以30%甲醇水溶液稀釋10倍后,在擬訂的分析條件下進樣10.0 μL。通過二極管陣列全掃描技術,按擬訂的色譜條件設定單通道多波長,獲得藥材供試品溶液的紫外色譜圖。比較紫外色譜圖和全掃描總離子流圖,二者基本對應。然而,紫外色譜圖中有個別吸收很大的峰,在相應的總離子流圖中強度卻較弱,提示這些化合物在擬訂的質譜條件下很難被離子化;此外,總離子流圖上有許多強度較大的峰,在相應的紫外色譜圖上卻沒有明顯的吸收,推測紅花藥材中含有大量的紫外吸收弱、易離子化的化合物。在獲得藥材供試品溶液全掃描總離子流圖的基礎上,利用離子阱質譜的步進式碰撞技術,通過改變電場梯度和能量梯度,獲得總離子流圖中各峰的多級碎片信息,通過對照并結合相關文獻[4-5]對各峰進行歸屬(見表1),定性分析了19種化學成分,其中1種為未知化學成分。
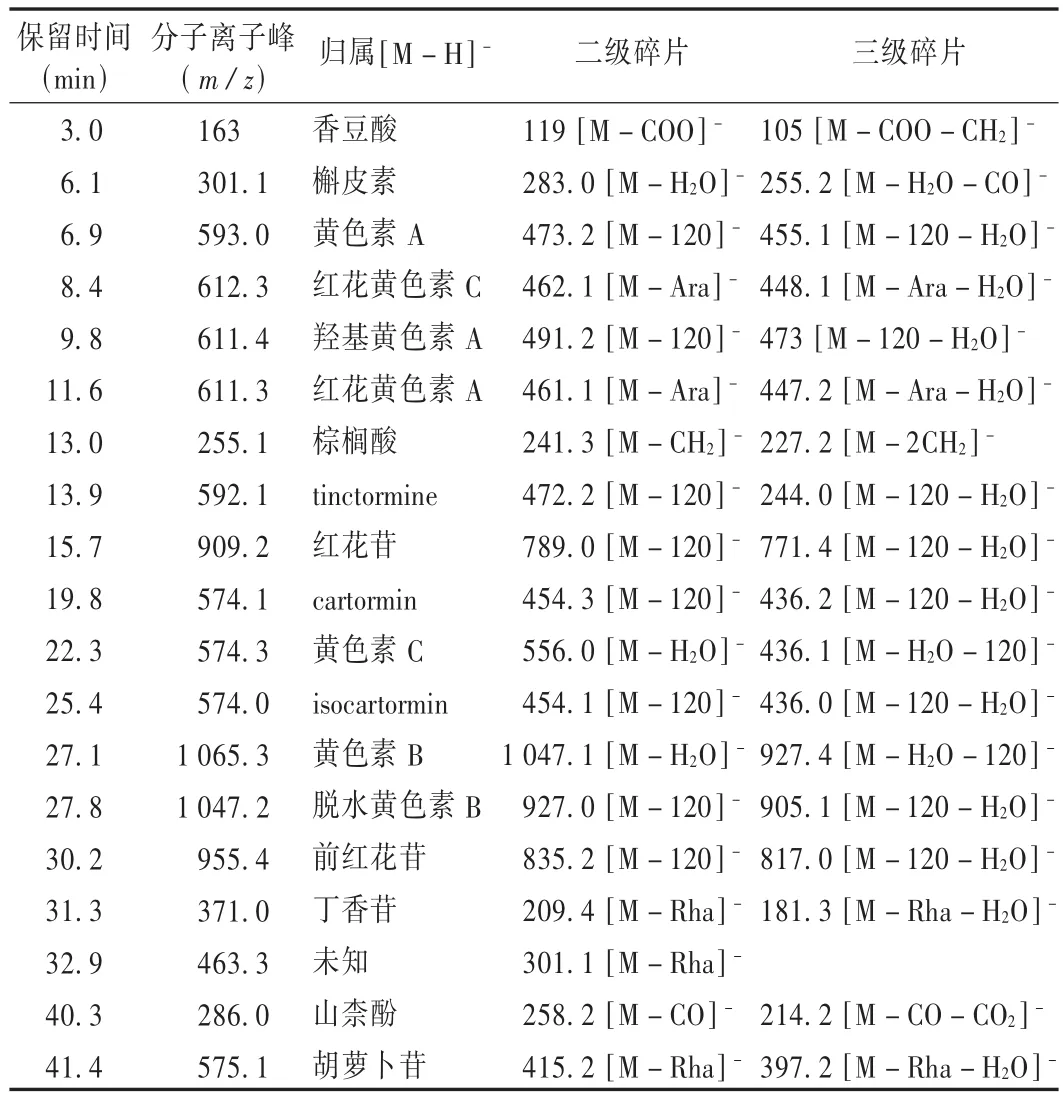
表1 紅花水提物電噴霧質譜解析表
3 討論
總結紅花中查耳酮類化合物在負離子模式下的裂解特征,發現該類化合物的結構中糖與苷元以C—苷鍵形式連接,通常情況下C—苷鍵較難發生斷裂。因此,在負離子模式下,[M-H]-離子主要發生3'位或4'位取代基上的消除反應,以延長苷元結構中的共軛體系,如常可見丟失中性片斷(相對分子質量為120)后所得基峰碎片離子。而對于多數糖苷類(包括黃酮苷類)化合物,在負離子模式下,主要發生糖苷鍵斷裂,產生丟失糖片斷后所得的碎片離子。該特性可用于區分醌型查耳酮類化合物與黃酮類化合物。
通過HPLC-ESI-MSn分析,對紅花水提物中19種化學成分進行質譜解析,在確定18種化學成分的基礎上,初步發現了1種未知的化學成分,對這種化學成分結構的定性還需進一步的研究。
[1]國家藥典委員會.中華人民共和國藥典(一部)[M].北京:化學工業出版社,2005:103-104.
[2]康 麗,顏曉燕,辛志偉.紅花的藥理作用研究進展[J].西南軍醫,2008,10(6):136-138.
[3]趙明波,鄧秀蘭,王亞玲,等.紅花RP-HPLC指紋圖譜的建立及其質量研究[J]. 藥學學報,2004,39(3):212-216.
[4]姜建雙,夏鵬飛,馮子明,等.紅花化學成分研究[J].中國中藥雜志,2008,33(24):2911-2913.
[5]周玉枝,陳 歡,喬 莉,等.紅花化學成分研究[J].中國藥物化學雜志,2007,17(6):380-382.
