癌復康膠囊Ⅱ號質量標準研究
2010-09-13 01:53:28孔俏玲李瑞明武漢大學武漢市40072廣東食品藥品職業學院廣州市510520中山大學附屬第一醫院藥學部廣州市510080
中國藥房 2010年39期
孔俏玲,李瑞明(1.武漢大學,武漢市 40072;2.廣東食品藥品職業學院,廣州市 510520;.中山大學附屬第一醫院藥學部,廣州市 510080)
癌復康膠囊Ⅱ號質量標準研究
孔俏玲1,2*,李瑞明3(1.武漢大學,武漢市 430072;2.廣東食品藥品職業學院,廣州市 510520;3.中山大學附屬第一醫院藥學部,廣州市 510080)
目的:建立癌復康膠囊Ⅱ號的質量標準。方法:采用薄層色譜(TLC)法對方中葛根、西洋參進行定性鑒別,采用高效液相色譜法對葛根素進行含量測定。結果:TLC斑點清晰、分離度好,陰性對照無干擾;葛根素檢測濃度在50.0~750.0 μg·mL-1范圍內與峰面積積分值呈良好的線性關系(r=0.999 9),平均回收率為99.51%,RSD=0.81%(n=6)。結論:定性定量方法操作簡便,結果準確、可靠,精密度好,所建標準可用于該制劑的質量控制。
癌復康膠囊Ⅱ號;質量標準;葛根素;薄層色譜法;高效液相色譜法
癌復康膠囊Ⅱ號是由當歸、升麻、葛根、苦參、西洋參等15味藥構成,具有獨特療效的醫院制劑。臨床研究發現,癌復康膠囊Ⅱ號具有活血抗癌的功效,可以提高人體機能,加快術后康復,減輕因放、化療帶來的副作用,在癌癥的輔助治療及術后康復等方面有滿意的臨床效果。筆者對本品進行新的質量標準的制定,增加葛根、西洋參2味藥材的薄層色譜(TLC)鑒別。同時,由于葛根為本品的主要藥味之一,葛根素(Puerarin)為葛根的主要成分之一[1],因此,本試驗采用高效液相色譜(HPLC)法測定癌復康膠囊Ⅱ號中葛根素含量,以有效控制產品的內在質量,提高臨床療效。
1 儀器與材料
1.1 儀器
L114型萬分之一電子分析天平(瑞士Mettler Toledo公司);CQ-25型超聲波儀(上海超聲波儀器廠);硅膠G、H薄層板(青島海洋化工廠);2100型紫外-可見分光光度計及HPLC儀,包括SPD-10Avp紫外檢測器、LC-10ATvp泵、N-2000雙管道色譜數據工作站(日本島津公司)。
1.2 試藥
葛根素對照品,葛根、西洋參對照藥材(中國藥品生物制品檢定所,批號分別為0125-200207、1175-200501、120997-200508);癌復康膠囊Ⅱ號樣品及陰性樣品均由天津中醫學院第二附屬醫院制劑室自制;水為2次重蒸水,其余試劑均為分析純。
2 定性鑒別
2.1 葛根的TLC鑒別[2,3]
取本品內容物粉末6 g,加甲醇30 mL,超聲1 h,濾過,濾液蒸干,殘渣加甲醇1 mL使溶解,作為供試品溶液;另取葛根對照藥材粉末6 g,同法制成對照藥材溶液;再取缺葛根陰性樣品粉末6 g,同法制成陰性對照溶液。照TLC法[2]試驗,吸取上述3種溶液各6 μL,分別點于同一硅膠H薄層板上,使成條狀,以氯仿-甲醇-水(5∶1∶0.1)為展開劑,展開,展距為13 cm,取出,晾干,紫外光燈(365 nm)下檢視。結果,供試品色譜中,在與對照藥材色譜相應的位置上,顯相同顏色的熒光條斑;陰性對照無干擾。葛根的TLC見圖1。
2.2 西洋參的TLC鑒別[2]
取本品內容物粉末3 g,加甲醇25 mL,加熱回流1 h,放冷,濾過,濾液蒸干,殘渣加水20 mL使溶解,乙醚振搖提取2次,每次10 mL,水層再用水飽和正丁醇振搖提取3次,每次15 mL,合并飽和正丁醇溶液,用水洗滌2次,每次10 mL,再用稀氨水10 mL洗滌1次,棄去水層,飽和正丁醇溶液蒸干,殘渣加甲醇1 mL使溶解,作為供試品溶液;另取西洋參對照藥材粉末1 g,同法制成對照藥材溶液;再取缺西洋參陰性樣品粉末3 g,同法制成陰性對照溶液。照TLC法[2]試驗,吸取上述供試品溶液、陰性對照溶液各4 μL,對照藥材溶液2 μL,分別點于同一硅膠G薄層板上,使成條狀,以氯仿-甲醇-水(69∶27∶4)為展開劑,展開,展距為12 cm,取出,晾干,噴以10%硫酸乙醇溶液,于105℃加熱至斑點清晰,紫外光燈(365 nm)下檢視。結果,供試品色譜中,在與對照藥材色譜相應的位置上,分別顯相同顏色的斑點或熒光斑點;陰性對照無干擾。西洋參的TLC見圖2。
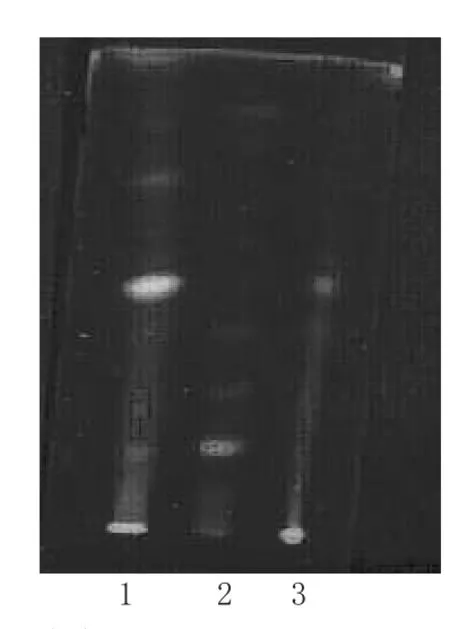
圖1 葛根的TLC1.供試品;2.葛根對照藥材;3.陰性對照Fig 1 TLC of Pueraria lobata1.test sample;2.Puerariae Lobatae Radix;3.negative control
3 葛根素的含量測定
3.1 色譜條件與系統適用性試驗
色譜柱:Thermo CTO-10A(250 mm×4.6 mm,5 μm);流動相:甲醇-水(25∶75);檢測波長:250 nm;流速:1.0 mL·min-1;進樣量:10 μL;柱溫:30 ℃;柱壓:106 Pa;靈敏度:0.01 AUFS。理論板數按葛根素色譜峰計算為5 340,其余組分的色譜峰能達到基線分離,分離度為1.9。供試品與葛根素對照品在相同時間點出現同一色譜峰,陰性對照未出現色譜峰。色譜見圖3。
3.2 溶液的制備[4]
3.2.1 對照品溶液的制備 準確稱取葛根素對照品3.1 mg,置于100 mL量瓶中,加50%甲醇水溶液溶解并稀釋至刻度,搖勻,得0.031 mg·mL-1的葛根素對照品溶液。

圖2 西洋參的TLC1.供試品;2.西洋參對照藥材;3.陰性對照Fig 2 TLC of Panax quinquefolium1.test sample;2.Panacis Quinquefolii Radix control;3.negative control
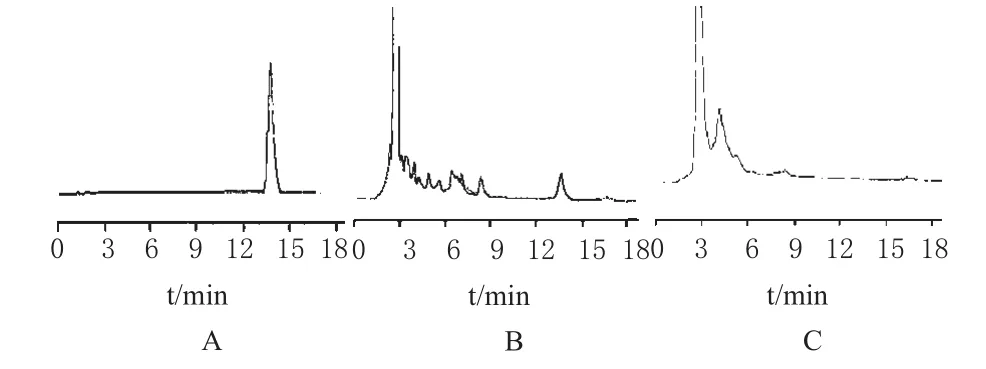
圖3 高效液相色譜圖A.供試品;B.葛根素對照品;C.陰性對照Fig 3HPLCA.test sample;B.Puerarin control;C.negative control
3.2.2 供試品溶液的制備 取樣品5粒,傾出內容物(約相當于含葛根素0.740 6 mg),置于50 mL量瓶中,加50%甲醇水溶液近刻度,超聲處理1 h,取出,放冷,再加50%甲醇水溶液至刻度,搖勻,用微孔濾膜(0.45 μm)濾過,取續濾液作為供試品溶液。
3.2.3 陰性對照溶液的制備 取本品缺葛根的陰性樣品粉末適量(約2 g),按“3.2.2”項下方法同法制成陰性對照溶液。
3.3 標準曲線的制備
精密吸取0.031 mg·mL-1的葛根素對照品溶液1、3、6、9、12、15 μL,按上述色譜條件進樣測定。以葛根素檢測濃度(X)為橫坐標,峰面積積分值(Y)為縱坐標,制備標準曲線,得回歸方程為Y=4 118.9X+404.12(r=0.999 9)。結果表明,葛根素檢測濃度在50.0~750.0 μg·mL-1范圍內與峰面積積分值呈良好線性關系。
3.4 精密度試驗
取同一批次(批號:050302)供試品溶液適量,按上述色譜條件進樣測定6次,記錄色譜圖。結果,RSD=2.18%(n=6),表明本方法精密度較好。
3.5 重復性試驗
分別取同一批(批號:050302)樣品6份,按“3.2.2”項下方法制備供試品溶液,照上述色譜條件測定,記錄色譜圖。結果,RSD=1.74%(n=6),表明方法重復性較好。
3.6 穩定性試驗
取供試品溶液(批號:050302),分別在0、2、4、6、8、10 h進樣,連續進樣6次,照上述色譜條件測定。結果,RSD=1.17%(n=6),表明供試品溶液在10 h內穩定。
3.7 加樣回收率試驗
精密稱取樣品(批號:050302)適量(共6份),分別置于50 mL量瓶中,各加0.03 mg·mL-1的葛根素對照品溶液10 mL,搖勻后再各加50%甲醇稀釋至刻度,搖勻,超聲1 h,再加50%甲醇至刻度,濾過,取續濾液,每份精密量取10μL進樣測定,計算加樣回收率,結果見表1。
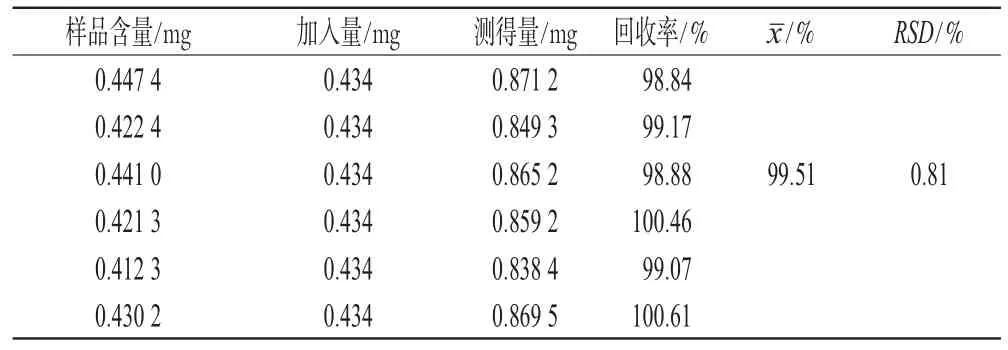
表1 加樣回收率試驗結果(n=6)Tab 1 Results of recovery test(n=6)
3.8 樣品含量測定
取5個批號的癌復康膠囊Ⅱ號,按“3.2.2”項下方法制備供試品溶液,照上述色譜條件測定,按外標法計算葛根素含量。結 果 ,5批(批 號 :0503022、0503023、0503024、0503025、0503026)樣品的含量分別為0.441 2、0.442 2、0.434 1、0.440 9、0.437 6 mg·mL-1,平均含量為0.439 2 mg·mL-1,RSD=1.42%(n=6)。
4 討論
葛根的TLC鑒別,把展開劑氯仿-甲醇-水的比例7∶2.5∶0.25[2]改為5∶1∶0.1,相應地降低了展開劑的極性,從而降低斑點隨展開劑移動的速度,達到更好的分離效果。
取0.05 mg·mL-1的葛根素對照品溶液進行紫外掃描。結果,葛根素在250 nm和306 nm波長處各有一個吸收峰。查閱資料可知,雖然選擇306 nm作為檢測波長時色譜圖中的組分峰會減少,色譜圖較為簡單,但選擇250 nm作為檢測波長,靈敏度最高,能有效檢測出葛根素特征峰,故確定選擇250 nm作為檢測波長。
[2,5,6],分別用 95%、30%、50%、25%的甲醇,超聲提取(20、30、60 min)樣品。結果,以50%甲醇超聲提取60 min的供試品溶液在甲醇-水(25∶75)為流動相的條件下,色譜效果最佳。較文獻[3]介紹的提取有效成分效率更高的熱回流法提取操作簡單、損失率低、主峰分離好。
根據2005年版《中國藥典》(一部)中HPLC法測定葛根素含量的方法,選擇甲醇-水(25∶75)為流動相。結果,葛根素的分離情況很好,保留時間較短,約為13 min;其余組分亦能在較短的時間內出峰,從而縮短了分析時間。
綜上,本試驗制定的方法操作簡便,結果準確、可靠,精密度好,所建標準可用于該制劑的質量控制。
參考文獻
[1]方建國,潘學暉,王文清,等.高效液相色譜法測定脈君安膠囊葛根素與氫氯噻嗪含量[J].醫藥導報,2008,27(5):592.
[2]國家藥典委員會編.中華人民共和國藥典(一部)[S].2005年版.北京:化學工業出版社,2005:附錄31、234、附錄313、87.
[3]彭菊艷,王俊儒,張義英,等.葛根有效成分的含量測定與提取工藝優化[J].陜西農業科學,2005,51(3):52.
[4]劉同祥,王 勇.HPLC測定消渴膠囊中葛根素的含量[J].云南中醫學院學報,2009,32(3):33.
[5]鄭國華,徐 晶,陶君彥,等.兒瀉停膠囊的質量標準研究[J].中醫藥學刊,2005,23(7):1 318.
[6]方繼輝,朱炳輝,鄒玉婷,等.復方葛根合劑及葛根中葛根素含量高效液相色譜分析[J].中國藥學雜志,2001,36(5):336.
Study on Quality Standard of AifukangⅡCapsules
KONG Qiao-ling(Wuhan University,Wuhan 430072,China)
KONG Qiao-ling(Guangdong Food and Drug Vocational College,Guangzhou 510520,China)
LI Rui-ming(Dept.of Pharmacy,The First Affiliated Hospital of Sun Yet-sen University,Guangzhou 510080,China)
OBJECTIVE:To establish the quality standard for AifukangⅡcapsules.METHODS:TLC was used for the qualitative identification ofPueraria lobataandPanax quinquefolium.HPLC was used to determine the content of puerarin.RESULTS:TLC spots were clear and well-separated without negative interference.The linear range of puerarin was 50.0~750.0 μg·mL-1(r=0.999 9)with an average recovery of 99.51%(RSD=0.81%,n=6).CONCLUSION:The method is simple,accurate and reliable with good precision.It can be used for the quality control of AifukangⅡcapsules.
AifukangⅡ capsules;Quality standard;Puerarin;TLC;HPLC
R283.65;R927.1
A
1001-0408(2010)39-3724-03
2010-04-02
2010-08-30)
