高效毛細管電泳同時分離多種酚類物質
2010-11-04 13:55:33周勝男檀華蓉李慧陸
中國糧油學報 2010年6期
周勝男檀華蓉李 慧陸 寧
(安徽農業大學茶與食品科技學院1,合肥 230036)
(安徽農業大學生物技術中心2,合肥 230036)
(安徽農業大學動物科技學院3,合肥 230036)
高效毛細管電泳同時分離多種酚類物質
周勝男1檀華蓉2李 慧3陸 寧1
(安徽農業大學茶與食品科技學院1,合肥 230036)
(安徽農業大學生物技術中心2,合肥 230036)
(安徽農業大學動物科技學院3,合肥 230036)
建立了高效毛細管電泳同時分離測定蘆丁、槲皮素、綠原酸、咖啡酸、沒食子酸和原兒茶酸 6種酚類物質的分析方法。考察了緩沖液種類、離子濃度和 pH、分離電壓、運行溫度等電泳參數,確立了最佳的電泳條件:緩沖液為 20 mmol/L磷酸二氫鈉 -20 mmol/L硼砂,pH7.5,分離電壓為 20 kV,運行溫度為 30℃,紫外檢測波長 214 nm。結果表明,6種物質在 12 min內得到完全分離,且各組分質量濃度與峰面積呈良好的線性關系,R2為 0.998 7~0.987 4。方法精密度試驗中,遷移時間的 RSD為 0.280%~0.425%,峰面積的 RDS為 5.228%~8.506%,回收率為 85.11%~101.98%。該法快速、簡便、準確,具有較高的靈敏度。
高效毛細管電泳 同時分離 酚酸類
酚類化合物屬于植物次生代謝產物,是多羥基酚類化合物的總稱。它存在于包括馬鈴薯等許多植物組織中[1]。按結構可分為酚酸類、類黃酮類及 1,2-二苯乙烯和木酚素類[2]。多酚類物質參與植物生長繁殖過程,協助植物防御病原、天敵等侵害。在防病保健方面,多酚化合物尤其是以綠原酸為主的酚酸類物質具有利膽保肝、抗病毒、抑制突變、抗腫瘤、抗氧化、抗動脈硬化、防治冠心病與中風等心腦血管疾病以及抗菌等多種生理功能[3]。隨著天然產物開發的逐漸興起,植物多酚以其在植物界分布的廣泛性、生理功能的多樣性以及來源豐富性等特點,逐漸成為當前研究的熱點,被形象地稱為“一座有待開發的金礦”[4]。
目前,在酚類物質的分離測定方面,國內外主要是經薄層分離后用紫外進行檢測的方法[5-7]。此方法涂層工序繁瑣,分離效果欠佳,且不利于大量樣品的檢測和自動化連續檢測。隨著科技的進步,也逐步采用高效液相色譜[8-9],GC-MS等方法,雖然自動化程度高,但儀器昂貴,測試成本高,分析時間相對較長。毛細管電泳是上世紀 80年代發展起來的分離分析技術[10-11],是一類以毛細管為分離通道、以高壓直流電場為驅動力的新型色譜分離技術。該方法具有分離效率高,分離速度快,分析化合物種類廣泛,重現性好、樣品和試劑用量少等優點[12],是一種高效的分離分析技術,相對成本較低,且是一種綠色環保的檢測方法,在分析領域引起極大關注,發展迅猛。國內外運用毛細管電泳對蘆丁,槲皮素,綠原酸等的分析較多[13-14],但對黃酮和多種酚、酸類的同時分離未見報道。本研究采用毛細管膠束電泳方法同時對蘆丁、槲皮素、綠原酸、咖啡酸、沒食子酸和原兒茶酸 6種酚類物質進行分離,主要研究緩沖液種類、離子濃度和 pH、分離電壓、運行溫度等電泳參數對分離效果的影響。
1 材料與方法
1.1 儀器
P/ACET MMDQ毛細管電泳儀 (配有二極管陣列PDA檢測器和色譜工作站):美國 Beckman公司;75 μm×65 cm未涂層熔融石英毛細管:河北永年與銳伴色譜器件有限公司;KQ-250DE數控超聲波清洗器:上海昆山市超聲儀器有限公司;0.45μm微孔濾膜:上海半島實業有限公司凈化器材廠。
1.2 藥品與試劑
蘆丁、槲皮素、綠原酸、原兒茶酸、沒食子酸、咖啡酸:中國藥品生物制品檢定所;四硼酸鈉 (硼砂)、磷酸二氫鈉、甲醇等分析純試劑。
1.3 溶液的配制
1.3.1 緩沖液的配制
準確稱取硼砂0.762 7 g,超純水定容至100 mL,制成 20 mmol/mL硼砂緩沖液;準確稱取磷酸二氫鈉0.312 0 g,超純水定容至 100 mL,制成 20 mmol/mL磷酸二氫鈉緩沖液;取兩種溶液混合,搖勻,pH計測定 pH值,分別制備 pH 7.0、7.5、8.0、8.5的緩沖溶液,用直徑 0.45μm濾膜過濾后,備 1用。
1.3.2 標樣的制備
準確配制各標準溶液:綠原酸0.5 mg/mL,咖啡酸1.25 mg/mL,蘆丁、槲皮素、沒食子酸和原兒茶酸各1.0 mg/mL。取上述質量濃度的綠原酸 0.7 mL,咖啡酸0.28 mL,蘆丁、槲皮素、沒食子酸和原兒茶酸各0.35 mL,混合配置成混合液 (各標品質量濃度為 147 μg/mL)。再將混合液用 80%甲醇倍倍稀釋(依次吸取前一混合液 100μL,加入100μL 80%甲醇),得到6個質量濃度梯度 (73.5、36.75、18.375、9.187 5、4.593、2.297μg/mL)的混合液,存放于冰箱中備用。
1.3.3 樣液的制備
市售馬鈴薯洗凈晾干,切塊于 55℃熱風干燥至干,粉碎過 60目篩,取適量馬鈴薯干粉加入 70%乙醇水溶劑,料液比為 1︰ 12,超聲提取 3次,每次 40 min。合并提取液濃縮,用直徑 0.45μm濾膜過濾,冰箱保存備用。
1.4 電泳條件
毛細管柱在每次使用前依次用 0.1 mol/L氫氧化鈉溶液、超純水和電泳緩沖液各沖洗 10 min,兩次進樣間用 0.1 mol/L NaOH、超純水和緩沖液分別沖洗毛細管 3、3、4 min,以保證其重現性。緩沖體系為20 mmol/L磷酸氫二鈉 -硼砂緩沖液,pH 7.5, 3447.5 Pa,壓力進樣 5 s,在電壓 20 kV、柱溫 30℃下分離,于 214 nm波長處檢測。
2 結果與討論
2.1 檢測波長的確定
如圖 1所示,經紫外檢測器在 190~400 nm波長范圍內掃描,綠原酸和咖啡酸的特征吸收峰為 300~360 nm,蘆丁、槲皮素、沒食子酸和原兒茶酸的特征吸收峰為 200~220 nm,6種物質在 214 nm處都有較強吸收,所以選定 214 nm為最佳檢測波長。
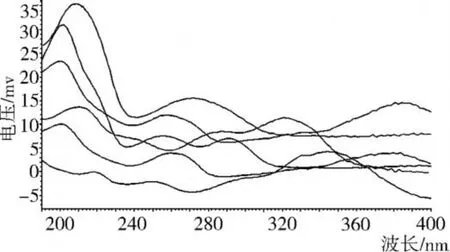
圖1 混合標樣的紫外特征吸收峰圖
2.2 緩沖體系的選擇
由于毛細管電泳的驅動力實際上是管內的電滲流[15],即毛細管中的溶劑因軸向直流電場作用而發生的定向流動。電滲流過大將引起樣品區帶變寬,降低分離效果。緩沖溶液的種類、濃度及體系 pH值對電滲流有直接影響,在同一緩沖體系中,電滲流將隨濃度的增加而降低。
2.2.1 電泳緩沖液種類的選擇
緩沖溶液背景電解質的選擇對毛細管區帶電泳分離效果有十分明顯的影響。如圖 2所示,本試驗在同一離子濃度 (20 mmol/L)和 pH(8.0)等條件下考察了 4種不同的緩沖體系:硼砂 -磷酸二氫鈉、硼砂 -硼酸、磷酸氫二鈉 -磷酸二氫鈉、磷酸氫二鈉 -硼酸緩沖體系。
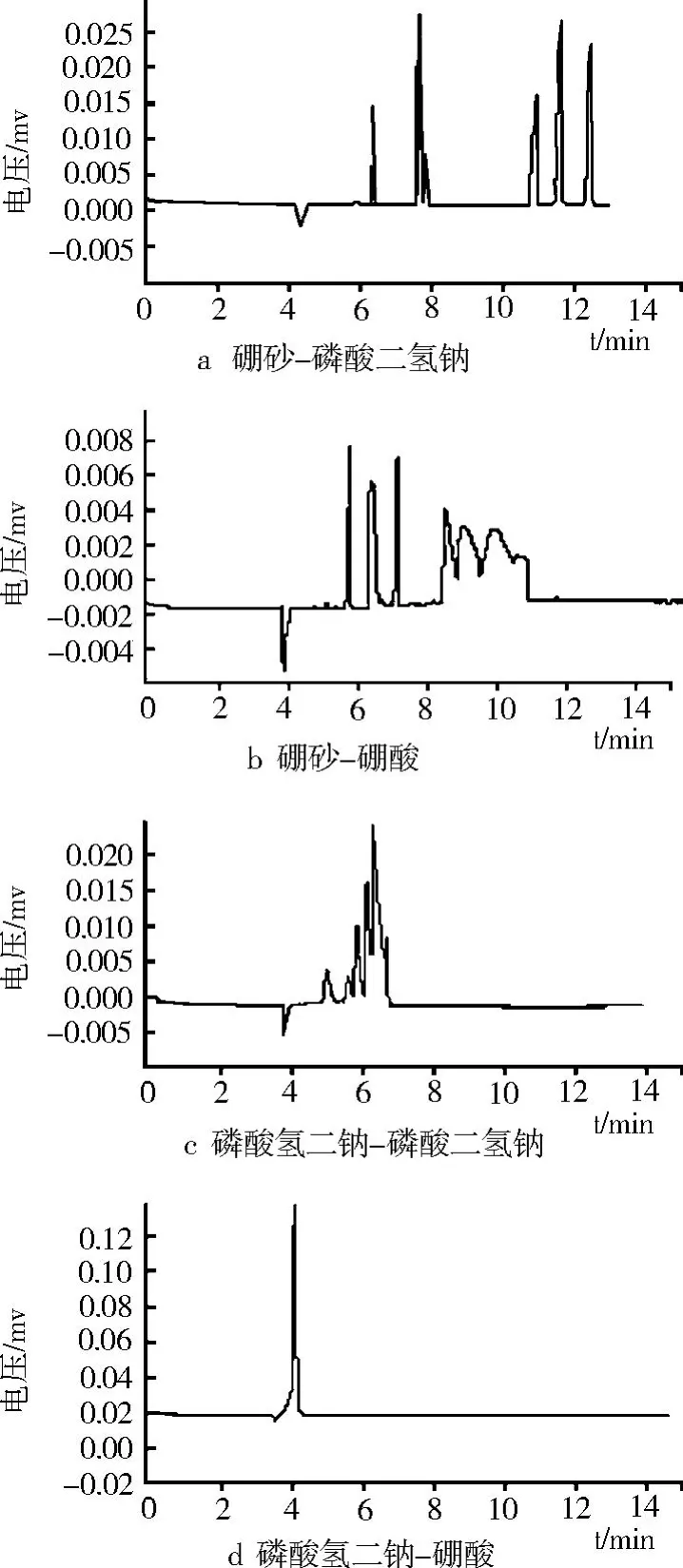
圖2 不同緩沖液類型的毛細管電泳圖譜
研究結果表明,硼砂 -磷酸二氫鈉緩沖液分離效果較好,峰形較佳;硼砂 -硼酸緩沖液體系前 3峰分離效果較好,但后 3峰峰形較差且峰拖尾明顯;磷酸氫二鈉 -磷酸二氫鈉緩沖體系分離效果較差,峰拖尾較嚴重;磷酸氫二鈉 -硼酸緩沖體系完全無法分離各組分。綜合考慮遷移時間與分離度的影響,硼砂 -磷酸二氫鈉緩沖液有較好的分離趨勢。因此,選擇硼砂 -磷酸二氫鈉作為試驗的運行緩沖液。
2.2.2 電泳緩沖液離子濃度的選擇
如圖 3所示,試驗過程中選擇了 20、30、40 mmol/L三個離子濃度的硼砂 -磷酸二氫鈉緩沖體系。試驗結果表明,緩沖液離子濃度對峰形和遷移時間有一定的影響,隨著離子濃度的增大,各組分的遷移時間延長,色譜峰變寬。當離子濃度為 20 mmol/L時,最后一個組分原兒茶酸的出峰時間 11. 988 min;而當離子濃度增大為 40 mmol/L時,最后一個組分原兒茶酸的出峰時間為 13.989 min。這可能由于緩沖液離子濃度增大導致溶液黏度增大,同時也引起毛細管內壁與溶液間的雙電層厚度減小,使電滲流減小,從而使各組分的遷移時間延長。綜合考慮遷移時間和峰形等因素,選擇 20 mmol/L作為分離的最佳離子濃度。
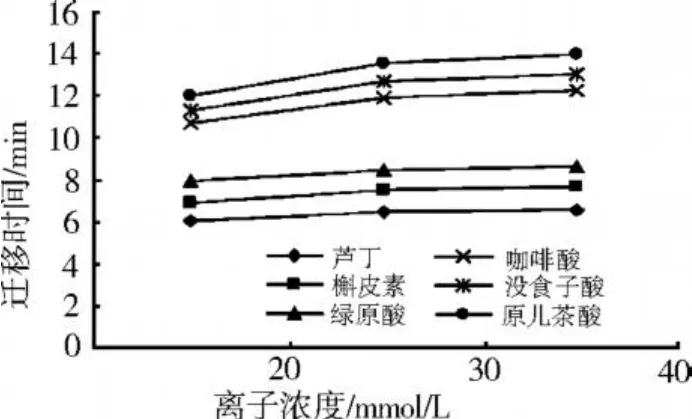
圖3 緩沖液離子濃度對遷移時間的影響
2.2.3 電泳緩沖液 pH的選擇
用毛細管電泳方法對物質的分離是因組分的離子淌度[16](所謂離子淌度,即指溶質離子在單位時間間隔內和單位電場下移動的距離)不同所致,離子的淌度與其有效電荷成正比,而離子的有效電荷易受操作緩沖溶液 pH值的影響。因而,緩沖溶液 pH值的調節與控制是優化分離的重要因素。
如圖 4所示,在 20 mmol/L硼砂 -20 mmol/L Na2HPO4緩沖液條件下,分別調至 pH7.0、7.5、8.0、8.5。研究結果表明,隨 pH的升高,毛細管內層表面的負電荷減少,電滲流減小,遷移時間相應延長。當pH 7.0時,后 3峰分離度較低,分離效果不好;當 pH 8.0時,槲皮素和綠原酸的分離效果較差;而當 pH 8.5時,雖然分離度較好,但遷移時間較長。綜合考慮遷移時間和各組分的分離度,選定 pH值為 7.5時分離效果較好。
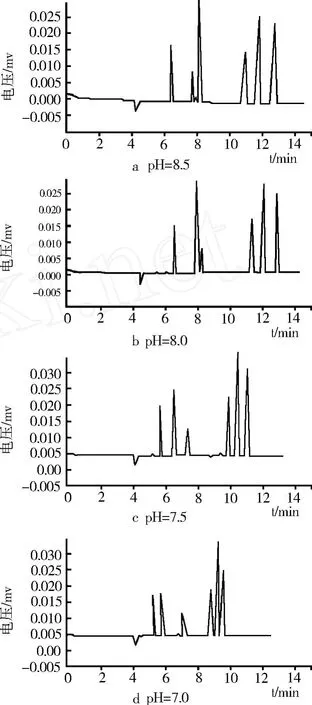
圖4 緩沖液pH對各組分分離度的影響
2.3 正交試驗
為了進一步優化最佳分離分析條件,在前面試驗的基礎上采用正交試驗設計,將緩沖體系的 pH、電泳的電壓和溫度作為3個因素,每個因素選取3個數量位級(水平),即三因素三水平試驗確定緩沖體系(如表1所示)。

表1 正交試驗因素水平表
由于毛細管電泳具有能在較短時間內達到最佳分離效果的優點,根據正交設計表L9(34)的各因素水平配置的方案(見表 2),本試驗分析了各組試驗條件下,完成分離的遷移時間及兩個色譜峰之間的最低分離度。
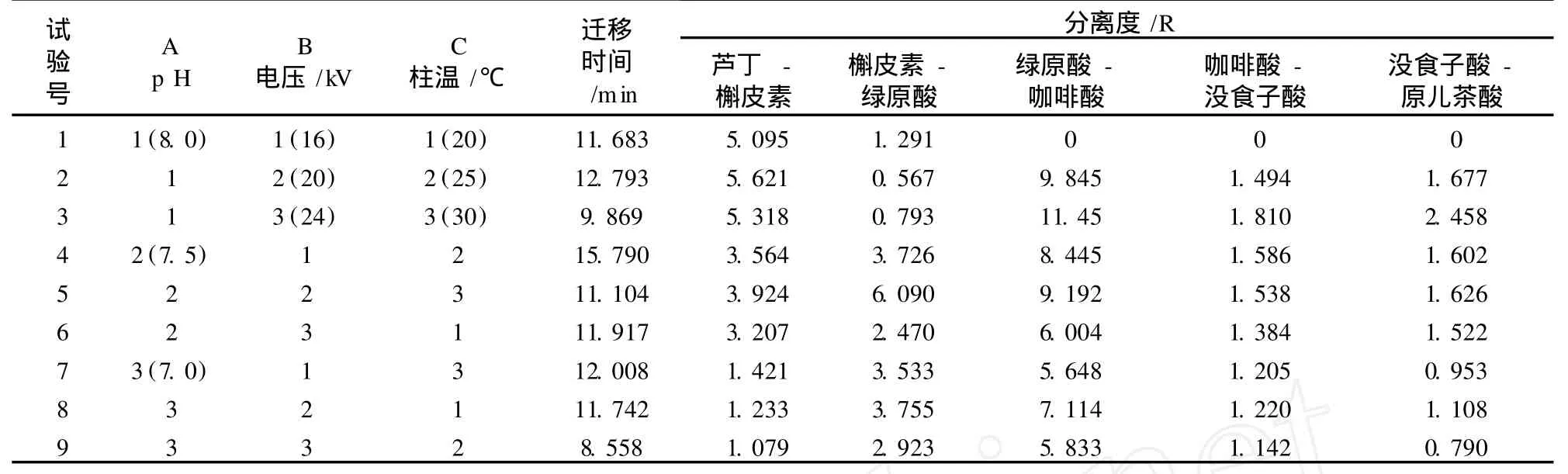
表 2 正交試驗表(L934)
高效毛細管電泳測定多種物質的關鍵性分離指標是分離度 R。通常情況下,認為 R>1.5即完全分離。若所有分離度 R均大于 1.5,則遷移時間越短越好。標準混合液的毛細管電泳圖如圖 5所示,由分析結果可知,只有第 4組和第 5組試驗的分離度 R> 1.5,但第 4組的遷移時間較長。綜合考慮分離度、遷移時間的因素,選擇第 5組即 pH 7.5,電壓 20 kV,運行溫度 30℃作為最佳電泳分離條件。
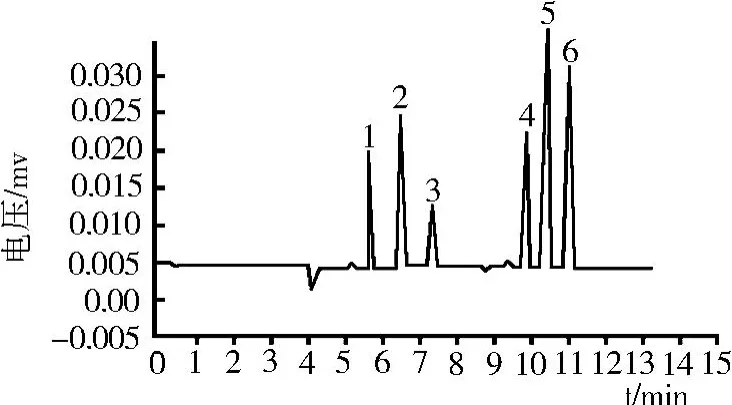
圖5 標準混合液的毛細管電泳圖
2.4 研究方法的評估
2.4.1 標準曲線及線性范圍確定
如表 3所示,在選定的條件下,以質量濃度由小到大的次序分別進樣 2.297、4.594、9.187 5、18.375、36.75、73.5μg/mL的標準工作液,按上述電泳條件進行分析,得到待測離子的峰面積,以質量濃度為橫坐標,以相應峰面積為縱坐標做回歸分析,得到標準曲線和回歸方程。分析結果表明,6組分線性關系良好,相關系數為 0.998 7~0.987 4。
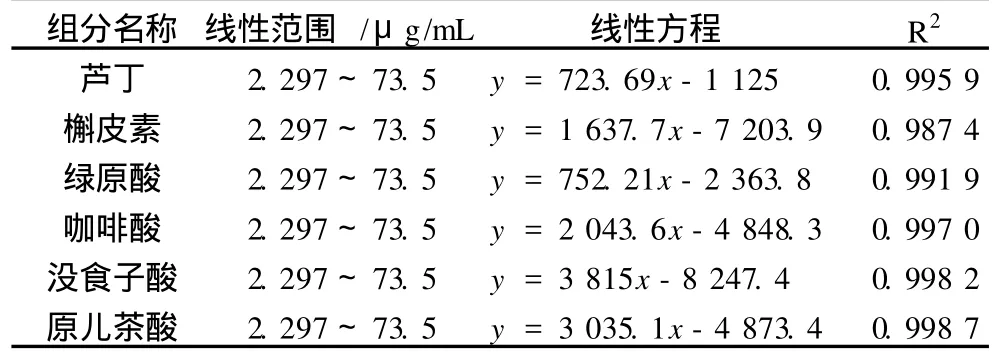
表 3 線性范圍和回歸方程
2.4.2 方法精密度研究
分析方法的重復性是評價其保持不受參數微小偏差影響的能力,可作為正常使用的一個可靠性指標。如表 4所示,在上述優化條件下 (即:緩沖液為20 mmol/L磷酸二氫鈉 -20 mmol/L硼砂,pH 7.5,分離電壓為 20 kV,運行溫度為 30℃,紫外檢測波長214 nm),取 36.75μg/mL混合標準溶液連續進樣 6次,考察各組分的遷移時間和校正峰面積的重復性。峰面積和遷移時間的相對標準偏差分別小于8.506%和 0.425%。
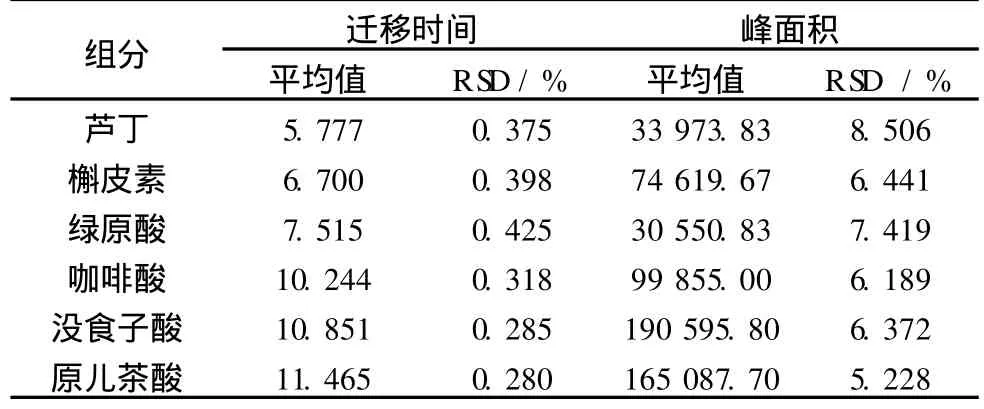
表 4 分析組分遷移時間和峰面積的精密度(n=6)
2.4.3 方法回收率研究
準確度驗證采用添加回收率法進行試驗。取100μL已知濃度的樣品三份,分別于其中加入 73.5、36.75、18.375μg/mL標準品混合溶液各 100μL,測定樣品。加標樣品的含量減去未加標樣品的含量即為實測添加量。實測添加量與標準添加量之比為回收率。通過試驗,測得蘆丁、槲皮素、綠原酸、咖啡酸、沒食子酸和原兒茶酸 6種酚類的回收率分別為97.73%、90.57%、93.60%、85.11%、100.17%、101.98%,RDS分別為 0.24%、0.11%、0.40%、0.19%、0.17%、0.16%,結果令人滿意。
2.5 樣品的測定
在最佳分離條件下,分離檢測樣品,計算酚酸類物質的含量(見圖 6)。從圖 6中可以很明顯的看出,馬鈴薯酚類物質主要為綠原酸和咖啡酸兩種。
馬鈴薯中酚類物質含量如表 5所示,其中綠原酸含量為 330.18μg/g,咖啡酸含量為 89.33μg/g,馬鈴薯中酚類物質總含量為 419.51μg/g。
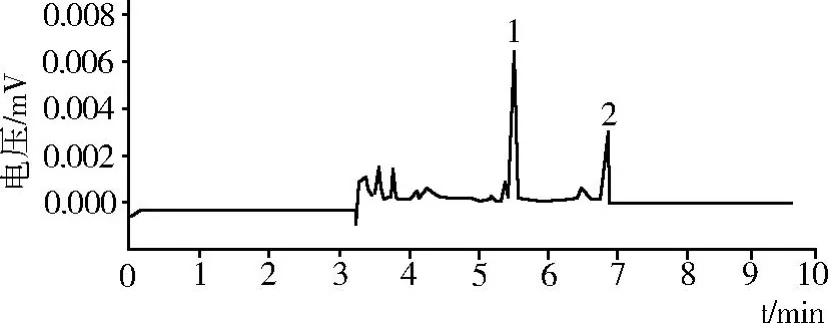
圖6 樣品液的毛細管電泳圖
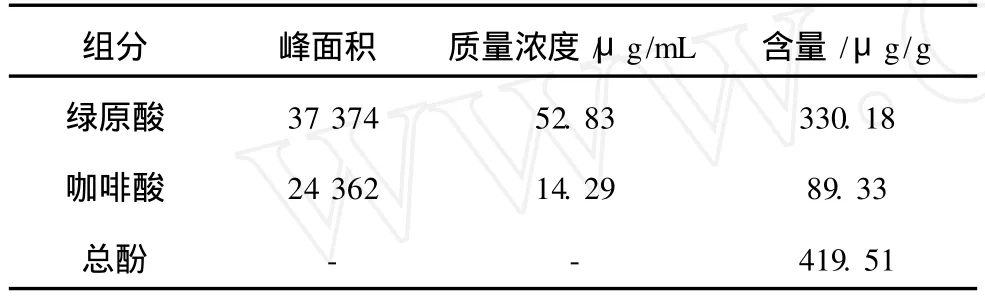
表5 酚類物質含量
3 結論
3.1 通過對電泳緩沖液類型、離子濃度和 pH值、分離電壓、分離溫度等電泳條件進行比較研究,確立了同時分離測定蘆丁、槲皮素、綠原酸、咖啡酸、沒食子酸和原兒茶酸 6種酚類物質的高效毛細管電泳方法,6種物質可在 12 min內得到完全分離。
3.2 研究并確立了最佳的電泳條件:緩沖液采用20 mmol/L磷酸二氫鈉 -20 mmol/L硼砂,pH 7.5,分離電壓 20 kV,運行溫度 30℃,紫外檢測波長214 nm。
3.3 本方法工作曲線線性關系良好、結果的準確度和精密度高,樣品用量少,分析時間短,與其他分析方法相比較,高效毛細管電泳法操作更簡便、快速,可在 12 min內同時分離 6種物質,大大減少了分析成本,為同時分離檢定多種酚類物質提供了一個良好的方法。
[1]Deshpande S S,Sathe S K,Salunkhe D K.Chemistry and Safety of plant Polyphenols[J].In Nutritional and Toxico2 logicalAspects of Food Safety.Friedman M,Ed,plenum: NewYork,1984:457-495
[2]郭長江,韋京豫.食材新寵兒多酚類物質 [J].中國食品報,2004,3:40-41
[3]馮麗,宋曙輝.植物多酚種類及其生理功能的研究進展[J].江西農業學報,2007,10:105-107
[4]韓丙軍,彭黎旭.植物多酚提取技術及開發現狀[J].華南熱帶農業大學學報,2005,1:21-24
[5]吳文標.馬鈴薯塊莖的 6-羥基 -7-甲氧基香豆素的分離和鑒定[J].馬鈴薯雜志,1999,4:79-80
[6]劉艷芳,馬歌麗.薄層色譜—分光光度法測定葵花籽中的綠原酸[J].現代食品科技,2007,11:80-82
[7]HE Zhao-fan,ZHANG Di-qing.Health food chemisty and detection technology[M].Beijing:Chinese Light Industry Press,1998:124-125
[8]劉麗梅,李曼玲.HPLC法測定秦皮中香豆素類成分的含量[J].中草藥,2004,7:819-822
[9]SylvesterN.Onyeneho andNavam S.Hettiarachchy.Antiox2 idant activityof durum wheat bran[J].JournalofAgricultur2 al and Food Chemistry 1992,40:1496-1500
[10]魏偉,王義明,羅國安.中成藥成分的高效毛細管電泳分析[J].藥學學報,1997,6:476-480
[11]王德先,趙敬湘,楊更亮等.毛細管區帶電泳法測定中藥金銀花中綠原酸的含量[J].中草藥,2000,6:432-433
[12]ZhimingLiu,Tao Li,Jie Li,et al.Detection ofmenadione sodium bisulfite(vitam in K3)by reversed-phase high perfor mance liquid chromatography with series dual-elec2 trode amperometric detector[J].Analytica Chi mica Acta, 1997,338:57-62
[13]VaherM,KoelM.Separation of polyhenolic compounds ex2 tracted from plant matrices using capillary electrophoresis [J].Journal of ChromatographyA,2003,990(1-2):225 -230
[14]Wang DX,Yang GL,Engelhardt H,et a1.Separation by capillary zone electrophoresis of the active anthraquinone components of the Chinese herb Polygonum multiflorum Thunb[J].Chromatographia,2001,53:185-186
[15]陳義.毛細管電泳技術及應用[M].北京:化學工業出版社,2000:14-15
[16]沈曉春,陳平,郭偉強,等.用毛細管電泳方法分離有機酸的研究[J].浙江大學學報:理學版,2002,29(6):679 -684.
Simultaneous Separation of Polyphenols by High Perfor mance Capillary Electrophoresis
Zhou Shengnan1Tan Huarong2Li Hui3Lu Ning1
(College of Tea and Food Seience and Techaology,AnhuiAgriculturalUniversity1,Hefei 230036)
(Biotechnology Center,AnhuiAgriculturalUniversity2,Hefei 230036)
(College ofAnimal Science,AnhuiAgriculturalUniversity3,Hefei 230036)
A high perfor mance capillary electrophoresismethod has been developed for the simultaneous sepa2 ration and deter mination of six phenolics of rutin、quercetin、chlorogenic acid、caffeic acid、gallic acid and protocate2 chuic acid.The effects of several factors such as the pH and concentration of running buffer,the separation voltage and the runnion tamperation on HPCE were investigated.Under optimum condition,the running buffer consisted of 20 mmol/LNaH2PO4-20 mmol/L sodium borate,pH 7.5,separation voltageat 20 kV,running temperation at 30℃, the UV detection wavelength was 214 nm.The results showed that the six analytes can be well separated within 12. 0 min,each one had a good linear relationship(R2=0.998 7~0.987 4).The RSD ofmigration time was 0.280%~0.425%and of the peak area was 5.228%~8.506%.The average recovery percentage was 85.11%~101.98%. The method is rapid,simple and accurate.
high herfor mance capillary electrophoresis,simultaneous separation,phenolic acids
O657.8;TS215 文獻標識碼:A 文章編號:1003-0174(2010)06-0114-06
2009-07-28
周勝男,女,1985年出生,碩士,農產品加工與貯藏
陸寧,女,1964年出生,教授,碩士生導師,食品及農產品貯藏與加工
