
羥基加成反應對A-T堿基對結構和質子轉移過程的影響
2010-11-30 10:49:18史俊友董麗花劉永軍
物理化學學報 2010年12期
史俊友 董麗花 劉永軍
(1中國科學院西北高原生物研究所,西寧 810001;2中國科學院研究生院,北京 100049)
羥基加成反應對A-T堿基對結構和質子轉移過程的影響
史俊友1,2,*董麗花1,2劉永軍1
(1中國科學院西北高原生物研究所,西寧 810001;2中國科學院研究生院,北京 100049)
應用密度泛函理論在B3LYP/6-31++G(d,p)//B3LYP/6-31G(d,p)水平上對羥基化堿基對A-T的結構進行了研究,經計算共得到8種穩定的羥基化加成產物,其能量的相對順序為8OHA-T<A-T6OH<A-T5OH<2OHA-T<4OHA-T<5OHA-T<A-T2OH<A-T4OH(數字表示羥基加成進攻的原子在腺嘌呤或胸腺嘧啶中的編號),這與其加成反應前后結構變化的大小密切相關.當羥基對腺嘌呤端進行加成時,A-T間的相互作用能略有增加,而當羥基對胸腺嘧啶進行加成時,A-T之間的相互作用能略有減小.另外,還以能量較低的加成產物8OHA-T和A-T6OH為例對羥基化堿基對中A和T之間的質子轉移過程進行了研究,結果表明羥基化產物中A與T之間的質子轉移機理由未加成前的分步雙質子轉移變為協同雙質子轉移,且其勢壘低于未加成的A-T發生第一步(決速步驟)質子轉移的勢壘.
密度泛函理論;堿基對;羥基化;質子轉移;單電子占據軌道
在DNA的雙螺旋結構中,各個堿基之間通過氫鍵相互作用以A-T(腺嘌呤-胸腺嘧啶)或G-C(鳥嘌呤-胞嘧啶)的方式進行配對.由于受環境因素的影響,這些堿基對之間的結合能力可能會發生變化,甚至誘發突變.生物體系內的水分子經紫外線輻射或細胞的正常代謝后可分解產生活性基團如H·、等.這些活性基團可與核苷作用導致DNA發生一系列的變化,如單、雙螺旋鏈結構的變化,堿基對間的錯配,突變等.由于這些自由基與核苷的反應具有嚴重的破壞性,其反應機理已經受到人們的密切關注.
人們通過實驗及理論研究發現·OH與DNA中的嘧啶(胸腺嘧啶或胞嘧啶)及嘌呤(腺嘌呤或鳥嘌呤)發生加成反應,且其相關產物已得到分離和鑒定[1-2].·OH與嘧啶的加成反應主要發生在C5及C6位,其中C5位的加成產物具有還原性,C6位的加成產物具有氧化性;·OH與嘌呤的加成反應可發生在C2、C4、C5、C8等位置,其產物中氧化性和還原性物質呈等量關系[3-5].這些加成產物仍比較活潑,不但可與其它生物分子發生反應,還可發生分子內的開環反應、分子內及分子間的抽氫反應等[5-6].通常情況下,核酸中的嘌呤和嘧啶主要以氨基和酮類的形式通過分子間氫鍵進行配對,但在少數情況下,嘌呤和嘧啶也以亞氨基和烯醇的形式存在(圖1).后一種情況的發生是由于嘌呤與嘧啶之間發生了質子轉移[7-9];它所占比例雖然很小,卻可以誘發基因突變,改變遺傳密碼[10].
為探討堿基對之間的質子轉移及其在基因突變中的作用,人們開展了許多相關的實驗及理論研究.結果表明G-C間的質子轉移遵循協同雙質子轉移機理[11],但對于堿基對A-T間的質子轉移機理,人們得到的結論并不完全一致,如Floribn[12]和Gorb[13]等認為堿基對A-T間的質子轉移機理為協同雙質子轉移,而近期Kryachk等[14]和Villani[15]的研究發現A-T間的質子轉移為分步質子轉移.在分步質子轉移中,首先胸腺嘧啶的H8由N3轉移到腺嘌呤的N1上,形成離子型中間體A-T',然后腺嘌呤N10上的氫原子轉移到胸腺嘧啶的O9上形成A'T'(圖1); Kryachk等[14]在HF/6-31+g(d)水平上計算得到的分步質子轉移的能壘分別為70.26和37.22 kJ·mol-1.
活性自由基與堿基相互作用的研究引起了人們的極大關注[119],但由于這些自由基活性較高,采用實驗方法對其研究具有較大的局限性,于是人們采用多種理論方法對羥基與單個堿基的相互作用進行了較為深入的研究,但對于羥基與堿基對之間的相互作用及其對堿基對之間質子轉移過程影響的研究還相對較少.Zhang等[20]用密度泛函方法研究了羥基自由基與G-C堿基對的加成反應及堿基之間的質子轉移過程,他們發現羥基加成對于G與C之間的相互作用有輕微的影響,但加成位置對G與C之間的質子轉移勢壘有不同的影響.到目前為止,對于羥基自由基與堿基對A-T之間的加成反應尚未見文獻報道.因此本文用密度泛函理論[21-22]對羥基加成后堿基對的結構變化以及A-T間的質子轉移過程進行了研究,以期加深對DNA損傷和修復過程的理解.
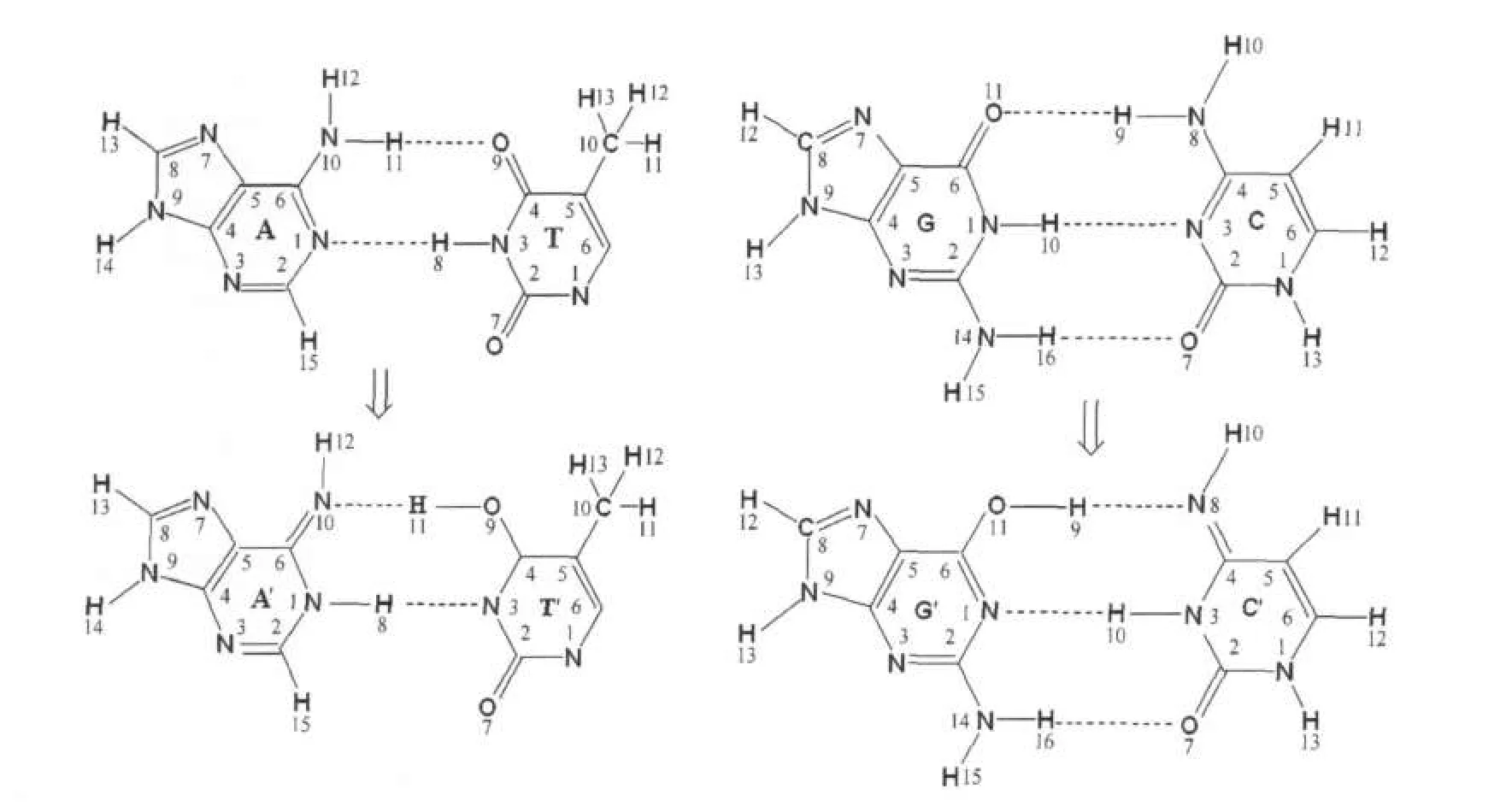
圖1 堿基對A-T和G-C及其質子轉移產物的原子編號標注示意圖Fig.1 Atom numbering scheme ofA-T and G-C base pairs and their proton transfer products
1 計算方法
因本文研究體系涉及到較多的氫鍵相互作用,我們采用已被證明對氫鍵體系十分有效的B3LYP方法[23-24]在6-31G(d,p)水平上對可能的羥基化產物及其質子轉移過程涉及到的各駐點的構型進行了全優化,并通過頻率計算加以確認;所有的局域最小點沒有虛頻,而相應的過渡態只有一個虛頻;過渡態還通過內稟反應坐標(IRC)加以確認,自然布居通過自然鍵軌道分析(NBO)得到.為使計算過程中所涉及的復合物、過渡態及產物的能量更加準確,在B3LYP/6-31++G(d,p)水平上對各優化構型進行了單點計算,下文中提到的能量都是指B3LYP/ 6-31++G(d,p)單點計算后所得到的能量(包含零點能校正).全部計算采用Gaussian 03程序包[25]完成.
為從本質上闡述A-T堿基對之間質子轉移勢壘變化的原因,本文還計算了有關羥基化堿基的質子親和勢(PA)與去質子焓(DPE).
對于質子化過程:B+H+→BH+
反應的吉布斯自由能變化通過下式計算:
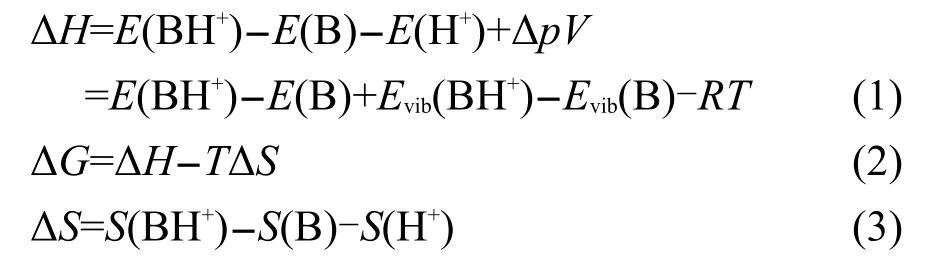
其中E(i)、Evib(i)和S(i)分別代表各物種i的總能量、零點振動能及熵;T和R分別為物質的絕對溫度及氣體常數;質子親和勢PA為吉布斯自由能變化(ΔG)的負值.對于質子化過程來說,PA的值越大,則該堿的堿性越強.
對于去質子化過程:BH→B-+H+
其焓變計算公式與式(1)相似:
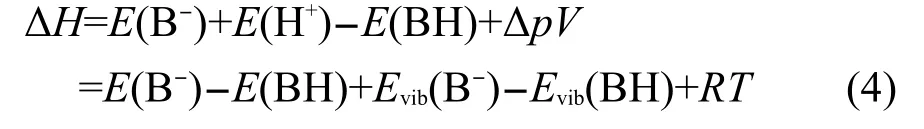
去質子焓DPE為該過程函變(ΔH)的負值.一般情況下,去質子焓越小表明該物質(BH)的酸性越強.
本文中所涉及的原子編號始終與圖1中A-T的編號一致.依據羥基加成位置的不同本文將·OH與A-T的加成產物命名為MOHA-T或A-TNOH,前者代表羥基加成到腺嘌呤上,后者代表羥基加成到胸腺嘧啶上,M和N分別代表羥基加成進攻的原子在腺嘌呤和胸腺嘧啶中的編號,MOHA-T(Ts/P)和A-TNOH(Ts/ P)分別代表質子轉移過程的過渡態或轉移后的產物.
2 結果與討論
2.1 加成產物的結構
對A-T堿基對中所有可能與·OH發生加成反應的不飽和原子進行了嘗試,包括腺嘌呤(A)中的2-8位原子,胸腺嘧啶(T)中的2-6位原子.經計算得到了8種穩定的加成產物,分別是2OHA-T、4OHA-T、5OHA-T、8OHA-T、A-T2OH、A-T4OH、A-T5OH和A-T6OH.
表1中列出了羥基氧原子與被加成原子所形成鍵的鍵長及相關原子上的電子自旋密度.可以看出,羥基氧原子上的電子自旋密度都很小,表明其單電子已經轉移到被加成堿基上;羥基與被加成原子形成了穩定的化學鍵,鍵長在0.1398-0.1445 nm之間.
表2列出了8種羥基化產物的總能量和相對能量,其中8OHA-T的能量最低,A-T4OH能量最高,其能量由低到高的順序為8OHA-T<A-T6OH<A-T5OH<2OHA-T<4OHA-T<5OHA-T<A-T2OH<A-T4OH.在腺嘌呤端的4種加成產物中,8OHA-T的能量最低,2OHA-T次之,而4OHA-T和5OHA-T的能量較高.在胸腺嘧啶端的4種產物中, A-T6OH的能量最低,A-T5OH次之;而A-T2OH與A-T4OH能量相當高,它們分別比8OHA-T的能量高144.42和153.20 kJ·mol-1,這與在溶液中未得到A-T2OH和A-T4OH的實驗結果[3-5]相一致.由于羥基的加成位置對產物的穩定性有較大的影響,我們對腺嘌呤和胸腺嘧啶加成產物的結構分別進行了討論.
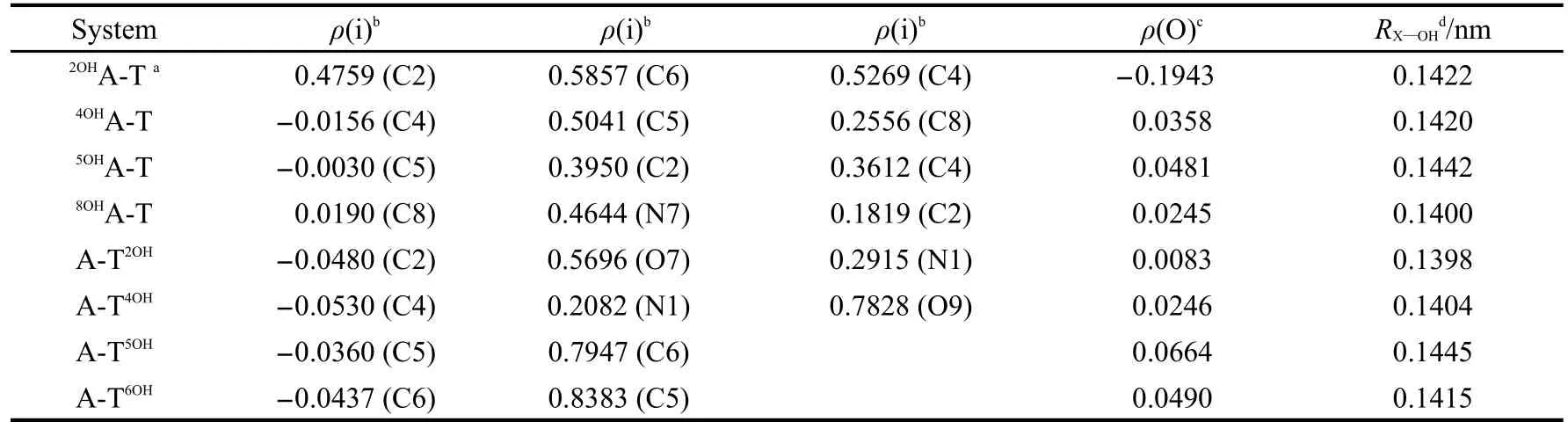
表1 羥基化堿基對中相關原子的電子自旋密度及X—OH的鍵長Table 1 Electron spin density of related atoms and bond length of X—OH for hydroxylated base pairs
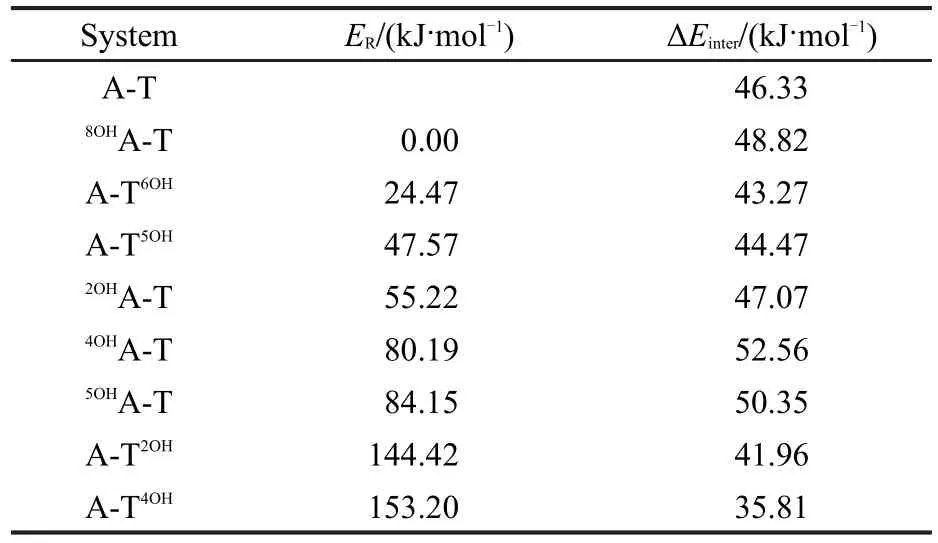
表2 羥基化堿基對A-T的相對能量(ER)及相應堿基間的相互作用能(ΔEinter)Table 2 Relative energies(ER)of hydroxylatedA-T and interaction energies(ΔEinter)between related bases
2.1.1 腺嘌呤端的加成產物
圖2列出了4種腺嘌呤端加成產物的結構.8OHA-T是·OH與腺嘌呤的C8加成反應的產物,其能量在·OH與腺嘌呤端的加成產物中最低.當·OH進攻C8時,C8的雜化方式由sp2轉化為sp3,C8與N7間的距離由0.1310 nm增長到0.1459 nm,即C8與N7間的化學鍵由雙鍵變為單鍵.在8OHA-T中,盡管C8上的氫原子向嘌呤環外發生扭曲,但嘌呤環的整體構型改變很小,仍基本保持Cs對稱性.2OHA-T的情況與8OHA-T類似,腺嘌呤部分的結構變化不大,基本保持Cs對稱性.而在4OHA-T和5OHA-T中,由于橋連碳原子C4和C5的雜化方式由sp2轉化為sp3,嘌呤環結構變化較大,五元環與六元環間呈蝶形扭曲.
羥基與腺嘌呤發生加成后,堿基對A-T間的氫鍵鍵長稍有變化.在8OHA-T、2OHA-T、4OHA-T和5OHA-T中,氫鍵N10—H11…O9較A-T(氫鍵為0.1930 nm)中分別縮短了0.0020、-0.0013、0.0030和0.0019 nm,氫鍵N3—H8…N1基本未變,其相應的相互作用能分別增加了2.49、0.74、6.23和4.02 kJ·mol-1(表2).這說明羥基對腺嘌呤C8、C2、C4和C5的加成對A-T間的氫鍵有增強作用.
2.1.2 胸腺嘧啶端的加成產物

圖2 B3LYP/6-31G(d,p)水平上優化得出的四種MOHA-T的相關構型Fig.2 Four structures ofMOHA-T optimized at the B3LYP/6-31G(d,p)levelThe unit of bond length in this figure and the following figures is nm.
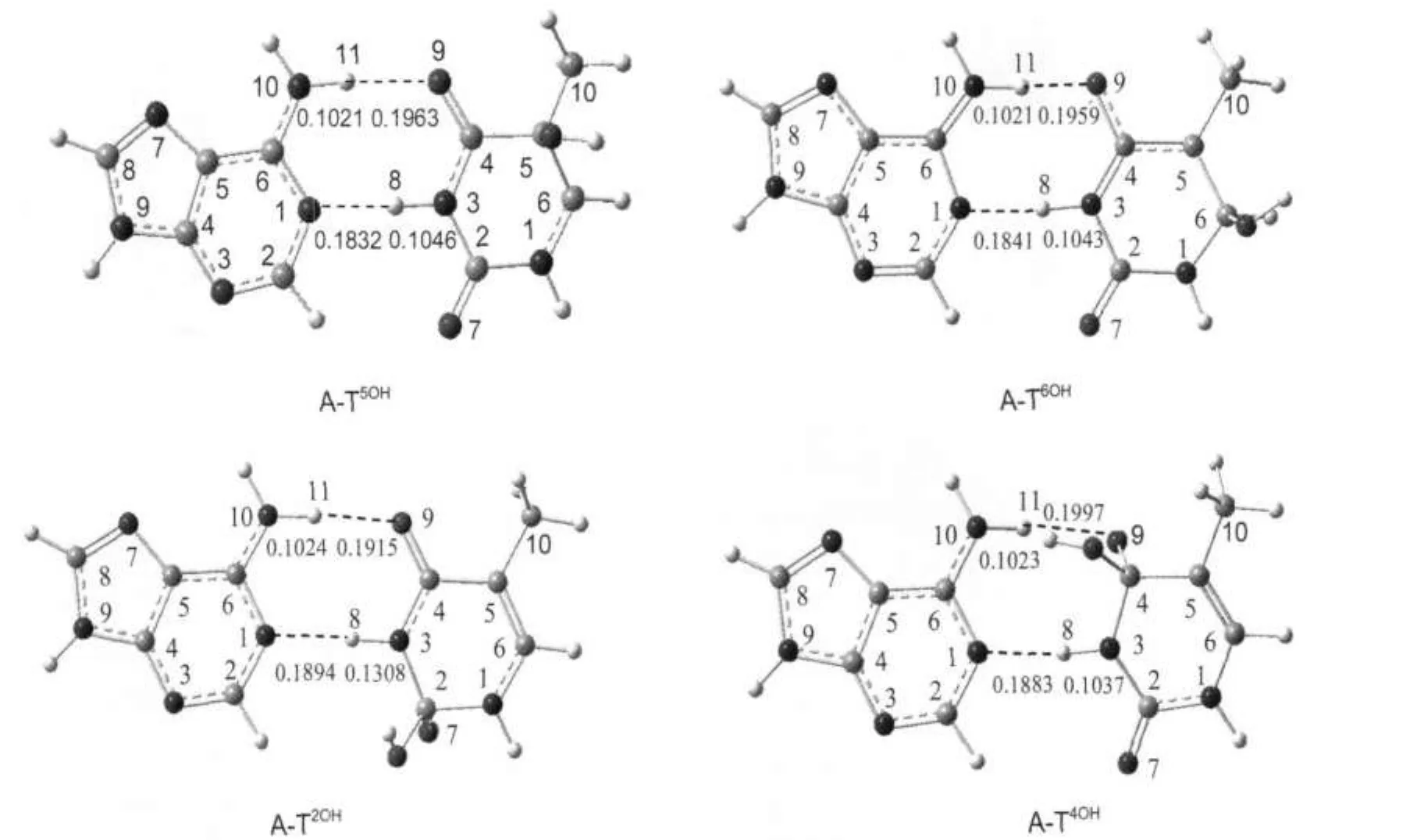
圖3 B3LYP/6-31G(d,p)水平上優化得出的四種A-TNOH的構型Fig.3 Four structures ofA-TNOHoptimizedat the B3LYP/6-31G(d,p)level
圖3為胸腺嘧啶端加成產物的結構.在A-T5OH的胸腺嘧啶端,C5的雜化方式由sp2轉化為sp3,C5與C6間的距離由0.1364 nm增長到0.1489 nm,二面角C2—N3—C4—C5與C2—N1—C6—C5分別由0°增加到10.67°和22.63°,表明C5已經脫離嘧啶環所在平面.而二面角N1—C2—N3—C4和C6—N1—C2—N3僅為4.32°和0.51°,表明胸腺嘧啶環中的其它五個原子仍基本保持共平面.A-T6OH與A-T5OH類似,C6脫離原嘧啶環所在平面,其它五個原子仍基本保持共平面,這說明A-T5OH與A-T6OH中的堿基T的結構變化較小.當羥基與胸腺嘧啶的C2與C4加成時引起堿基T的結構變化較大.在A-T2OH和A-T4OH中,C2與C4脫離嘧啶環所在的面,而O7與O9向面的另一側扭曲,其中C2—O7和C4—O9間的距離分別由0.1220和0.1235 nm增長到0.1323和0.1381 nm.在A-T4OH中,由于腺嘌呤中氨基與已扭曲的O9之間的氫鍵相互作用,導致嘌呤環和嘧啶環由共面變為異面,這可能導致DNA鏈的扭曲.
在A-T5OH、A-T6OH、A-T2OH和A-T4OH中,氫鍵N10—H11…O9較 在 A-T中 分 別 變 化 了 0.0033、0.0029、-0.0015和0.0067 nm,氫鍵N3—H8…N1的鍵長分別增長了0.0000、0.0009、0.0062和0.0051 nm,相應的相互作用能分別降低了1.86、3.06、4.37、10.52 kJ·mol-1.這表明羥基對胸腺嘧啶C5、C6、C2和C4的加成對A-T間的氫鍵有削弱作用.
通過分析8種加成產物的構型與能量發現,當加成產物的結構在A-T基礎上變化較大時,其能量較高,反之能量較低.當A發生羥基化時,堿基間的相互作用增強,而T發生羥基化時,相互作用減弱.
2.1.3 A-T羥基加成產物的電子結構
圖4列出了羥基與單個堿基和堿基對加合時產物的單電子占據軌道(SOMO)的能量值.與各單體加成產物的單電子占據軌道比較,堿基對A-T加成產物的單電子占據軌道能量普遍增高.復合物4OHA-T、5OHA-T及8OHA-T中單電子軌道的能量分別升高了0.003、0.006和0.005 a.u.,而2OHA-T的單電子軌道能量基本不變.A-T2OH、A-T4OH、A-T5OH和A-T6OH中的單電子占據軌道能量分別升高了0.032、0.010、 0.007和0.027 a.u..在腺嘌呤的四種加成產物中,5OHA-T的單電子軌道能量較高,易失去電子,其還原性較強.與之相反,2OHA-T和4OHA-T單電子軌道能量較低,易得到電子,具有較強的氧化性.胸腺嘧啶的四種加成產物中,A-T5OH的單電子占據軌道能量較高,具有較強的還原性;A-T2OH和A-T6OH的單電子占據軌道能量較低,氧化性較強,這與文獻報道的結果[16]一致.
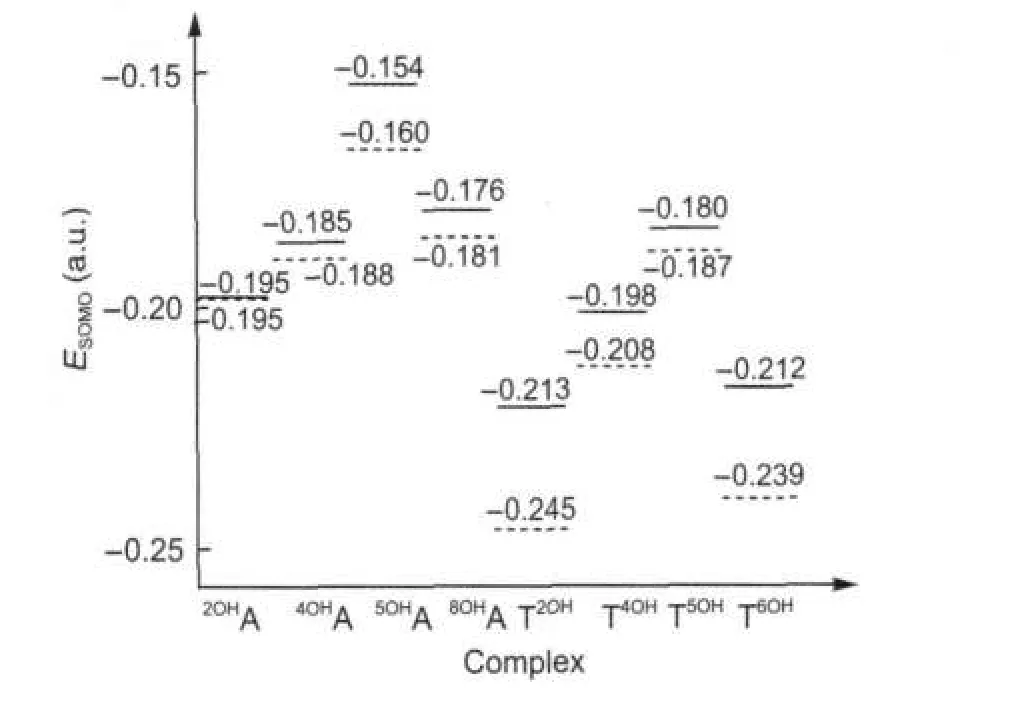
圖4 羥基化堿基單體OH-A/T(虛線)與相應的羥基化堿基對OH-A/T(實線)的單電子占據軌道(SOMO)的能量Fig.4 Singly occupied molecular orbital(SOMO) energies of monomers OH-A/T(dashed)and corresponding hydroxylation adducts OH-A/T(solid)
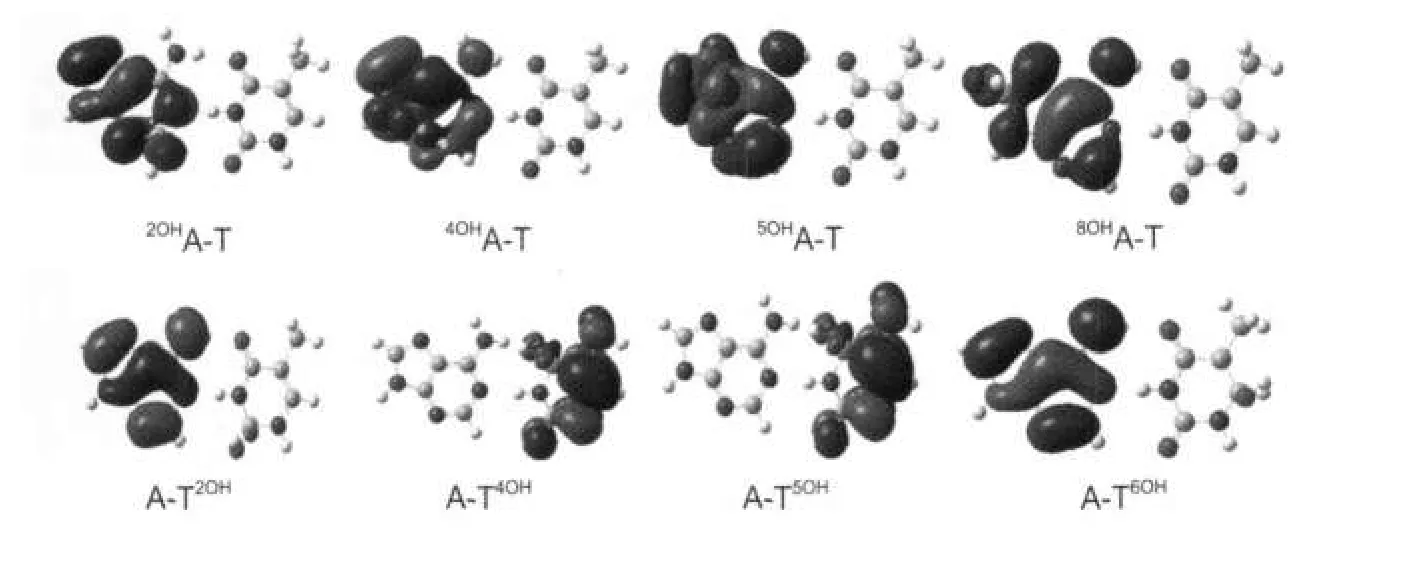
圖5 復合物MOHA-T和A-TNOH的單電子占據軌道圖Fig.5 SOMO populations of adductsMOHA-T andA-TNOH
圖5給出了8種羥基化產物SOMO的分布情況.當堿基對中的腺嘌呤羥基化加成時,SOMO主要分布在腺嘌呤一側;但是當羥基對胸腺嘧啶加成時,SOMO的分布情況與加成位置有很大關系,如 A-T4OH和A-T5OH的SOMO主要分布在胸腺嘧啶一側,而A-T2OH和A-T6OH的SOMO主要分布在腺嘌呤一側.2OHA、4OHA、5OHA和8OHA的SOMO的能量分別為-0.195、-0.188、-0.160和-0.181 a.u.,遠高于胸腺嘧啶的最高占據軌道(HOMO)的能量(-0.241a.u.),故當它們與胸腺嘧啶的HOMO作用時,SOMO主要分布在腺嘌呤一側.T2OH、T4OH、T5OH及T6OH的SOMO能量分別為-0.245、-0.208、-0.187和-0.239 a.u.,由于T4OH和T5OH的SOMO能量高于腺嘌呤的HOMO能量(-0.216 a.u.),故A-T4OH和A-T5OH的SOMO主要分布在胸腺嘧啶一側,而A-T2OH和A-T6OH的SOMO能量低于腺嘌呤的HOMO能量,因此它們的SOMO主要分布在腺嘌呤一側.
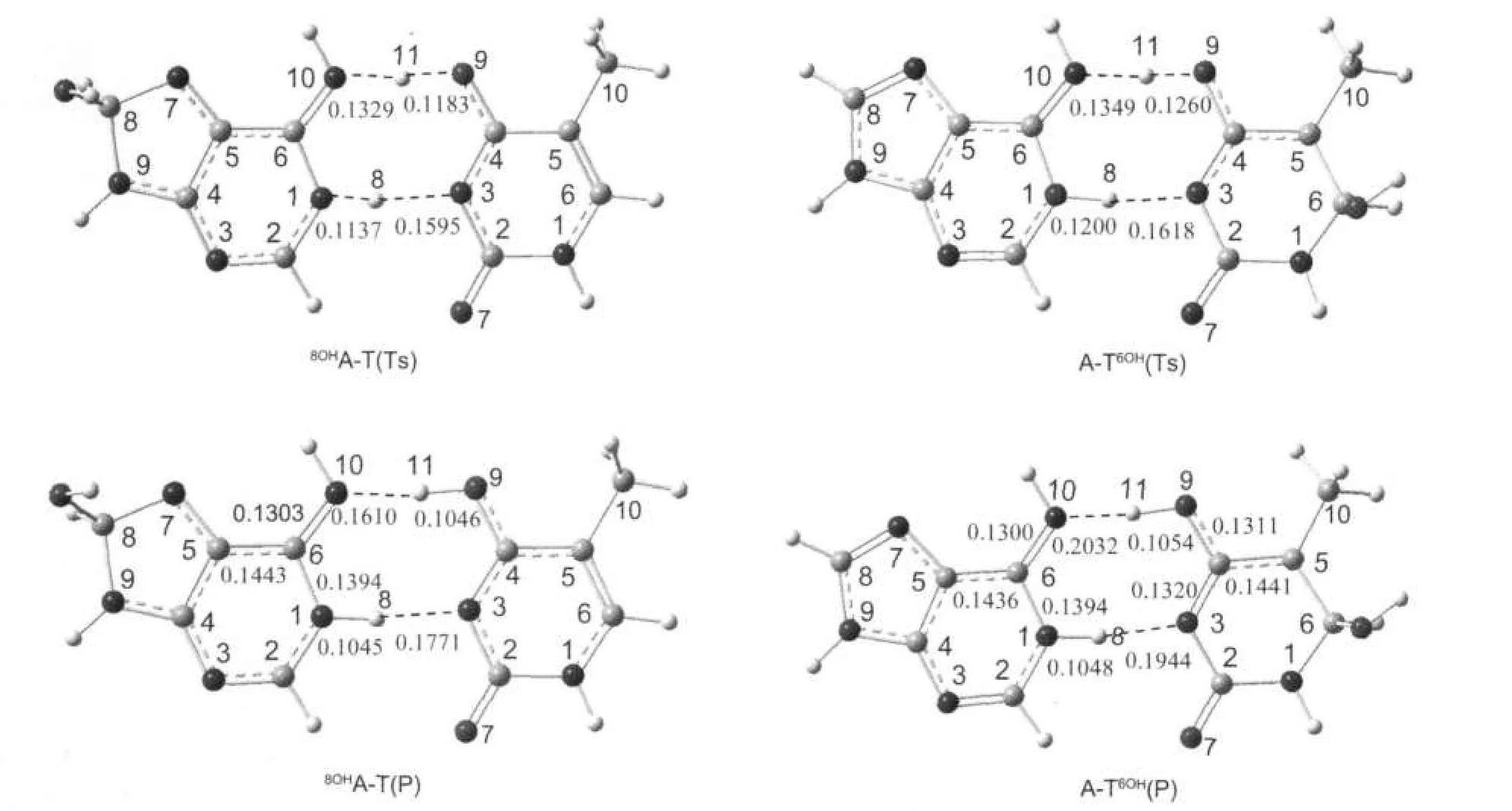
圖6 8OHA-T與A-T6OH質子轉移過程所涉及的過渡態與產物的優化構型Fig.6 Optimized structures of transition states and products of8OHA-T andA-T6OH corresponding to the proton transfer reaction

表3 8OHA-T與A-T6OH質子轉移過程所涉及的能壘和反應熱Table 3 Calculated activation barriers and reaction energies for the proton transfer reactions of 8OHA-T andA-T6OH
2.2 質子轉移的反應機理
本文以能量較低的兩種加成產物8OHA-T和A-T6OH為例對羥基化A-T間的質子轉移過程進行了研究.為便于比較,在B3LYP/6-31++G(d,p)//B3LYP/ 6-31G(d,p)水平上對未羥基化A-T間的質子轉移過程進行了研究,發現A-T間的質子轉移為分步質子轉移,其能壘分別為62.31和48.78 kJ·mol-1,與Kryachk等人的計算結果[14]基本一致.
圖6顯示了羥基化堿基對之間質子轉移的過渡態及產物的構型.表3中列出了8OHA-T與A-T6OH質子轉移過程所涉及的能壘和反應熱.我們對質子轉移過程中可能存在的離子型中間體進行了優化,但未能找到穩定的中間體,即整個質子轉移過程是協同進行的.本文對所得到的過渡態的結構進行了振動頻率計算,并通過IRC路徑分析予以確認.
計算表明羥基化A-T間的雙質子轉移發生在氫鍵N10(A)··O9(T)及N1(A)··N3(T)之間,H11由腺嘌呤轉移到胸腺嘧啶的O9上,同時胸腺嘧啶的H8向腺嘌呤的N1轉移.當8OHA-T和A-T6OH形成過渡態時過渡態的構型更接近產物.此結果符合Hammond假設[26],即若質子轉移的過程為吸熱過程,反應的過渡態與產物的構型更相近.
表3表明,8OHA-T和A-T6OH發生協同雙質子轉移時,其勢壘分別為42.01和50.27 kJ·mol-1,均低于A-T發生第一步質子轉移(決速步驟)的能壘,這說明羥基對于腺嘌呤的8號位和胸腺嘧啶的6號位的羥基加成有利于A-T間質子轉移的進行.另外,兩種加成產物質子轉移后的產物比質子轉移前的能量高48.34和55.67 kJ·mol-1,且與質子轉移過渡態的能量相差極小,所以它們質子轉移的產物極不穩定.8OHA-T和A-T6OH發生質子轉移的勢壘之所以比A-T發生第一步質子轉移的勢壘低,與堿基對中堿基酸堿性的變化有關.表4中列出了A/T及8OHA/T6OH中相關位置的質子親和勢與去質子焓.在8OHA中,8OHA端N10H11的去質子焓為1420.51 kJ·mol-1,與堿基A相比減小了25.92 kJ·mol-1,表明其酸性增強;N1位的質子親和勢僅減小了13.74 kJ·mol-1,這表明此位置的堿性基本不變.在T6OH中,N3位的去質子焓與T相比減小17.41 kJ·mol-1,酸性增強;但O9位置質子親和勢僅降低4.08 kJ·mol-1,堿性基本不變.在對應堿基不變的情況下,羥基化堿基的堿性基本不變,酸性增強,質子更易解離,故經羥基加成后A-T間的質子轉移勢壘降低.
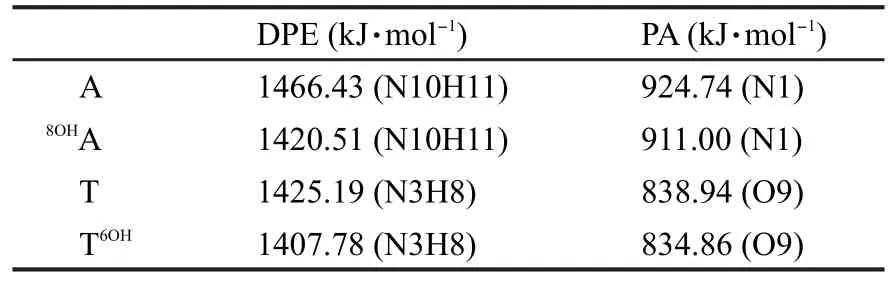
表4 A/T及8OHA/T6OH中相關位置的去質子焓與質子親和勢Table 4 Deprotonation enthalpies(DPE)and proton affinities(PA)for the relative positions ofA/T and8OHA/T6OH
3 結論
應用密度泛函理論在B3LYP/6-31G(d,p)水平上對羥基化堿基對A-T的結構進行了研究,并在B3LYP/6-31++G(d,p)水平上對相關結構進行單點能計算.經優化后共得到8種穩定的羥基化產物,產物能量的高低與加成發生前后的結構變化有密切關系,能量的相對順序為8OHA-T<A-T6OH<A-T5OH<2OHA-T<4OHA-T<5OHA-T<A-T2OH<A-T4OH.當羥基進攻腺嘌呤時,堿基對間的相互作用增加;而當羥基進攻胸腺嘧啶時,其相互作用減弱.在腺嘌呤端的4種加成產物中,2OHA-T和4OHA-T具有較強的氧化性,5OHA-T具有較強的還原性;在胸腺嘧啶端4種加成產物中,A-T5OH的還原性較強,A-T2OH和A-T4OH的氧化性較強;經羥基化后A-T的質子轉移機理由分步雙質子轉移變為協同雙質子轉移,且其勢壘低于A-T發生質子轉移的勢壘.
1 Breen,A.P.;Murphy,J.A.Free Radical Bio.Med.,1995,18: 1033
2 Burrows,C.J.;Muller,J.G.Chem.Rev.,1998,98:1109
3 Steenken,S.;Novais,H.M.J.Phys.Chem.,1987,91:426
4 Vieira,A.J.S.C.;Steenken,S.J.Am.Chem.Soc.,1990,112: 6986
5 Steenken,S.Chem.Rev.,1989,89:503
6 Colson,A.O.;Sevilla,M.D.J.Phys.Chem.,1996,100:4420
7 Lowdin,P.O.Adv.Quantum Chem.,1965,2:213
8 Piccirilli,J.A.;Krauch,T.;Moroney,S.E.;Benner,S.A.Nature, 1990,343:33
9 Benderskii,V.A.;Makarov,D.E.;Wight,C.A.Chemical dynamic at low temperature:advances in chemical physics.New York:Wiley&Sons,1994
10 Zhanpeisov,N.U.;Leszczynski,J.J.Phys.Chem.A,1998,102: 6167
11 Truhlar,D.G.Chem.Phys.Lett.,1998,294:45
12 Floribn,J.;Hrouda,V.;Pave1,H.J.Am.Chem.Soc.,1994,116: 1457
13 Gorb,L.;Podolyan,Y.;Dziekonski,P.;Sokalski,A.;Lexzczynski, J.J.Am.Chem.Soc.,2004,126:10119
14 Kryachk,E.S.;Sabin,J.R.Int.J.Quantum Chem.,2003,91:695
15 Villani,G.Chem.Phys.,2005,316:1
16 Xie,H.;Xia,F.;Cao,Z.J.Phys.Chem.A,2007,111:4384
17 Zhang,J.D.;Schaefee III,H.F.J.Chem.Theory Comput.,2007, 3:115
18 Grand,A.;Morell,C.;Labet,V.;Cadet,J.;Eriksson,L.A. J.Phys.Chem.A,2007,111:8968
19 Li,L.;Wang,H.;Niu,X.J.;Li,Z.H.Chem.J.Chin.Univ.,2009, 30:1596 [李 瀾,王 竑,牛曉娟,李宗和.高等學校化學學報,2009,30:1596]
20 Zhang,R.B.;Eriksson,L.A.J.Phys.Chem.B,2007,111:6571
21 Hohenberg,P.;Kohn,W.Phys.Rev.B,1964,136:864
22 Feller,D.J.Chem.Phys.,1990,93:579
23 Lee,C.;Yang,W.;Parr,R.G.Phys.Rev.B,1988,37:785
24 Becke,A.D.J.Chem.Phys.,1993,98:5648
25 Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03. Revision B.03.Pittsburgh,PA:Gaussian Inc.,2003
26 Hammond,G.S.J.Am.Chem.Soc.,1955,77:334
August 2,2010;Revised:September 13,2010;Published on Web:October 18,2010.
Effect of Hydroxylation on Structures and Proton Transfer of A-T Base Pairs
SHI Jun-You1,2,*DONG Li-Hua1,2LIU Yong-Jun1
(1Northwest Institute of Plateau Biology,Chinese Academy of Sciences,Xining 810001,P.R.China;
2Graduate University of Chinese Academy of Sciences,Beijing 100049,P.R.China)
The structures and proton transfer processes of hydroxylated A-T base pairs were theoretically studied at the B3LYP/6-31++G(d,p)//B3LYP/6-31G(d,p)level.Our calculations revealed that hydroxyl radical could react with A-T at different positions to form eight stable adducts.The order of these adducts in energy is8OHA-T<A-T6OH<A-T5OH<2OHA-T<4OHA-T<5OHA-T<A-T2OH<A-T4OH(the number denotes the label of the atom in the A/T which is attacked by hydroxyl),which relates well with their structural changes upon the addition of hydroxyl radical.The interaction energy between A and T would increase slightly when hydroxyl radical reacts with the adenine,but it would decrease when the radical reacts with thymine. To study the proton transfer processes of the hydroxylated A-T base pairs,the most stable adducts,8OHA-T and A-T6OH,were selected to give calculations.The calculated results indicate that the proton transfer processes of8OHA-T and A-T6OHfollow the concerted mechanism,which is different from the stepwise mechanism of A-T.What is more,its energy barrier is lower than the corresponding energy of the latter's first step(rate-determining step).
Density functional theory;Base pair;Hydroxylation;Proton transfer;Singly occupied molecular orbital
O641
?Corresponding author.Email:shijunyou123@126.com;Tel:+86-15809781546.
The project was supported by the Program of“One Hundred Talents Program”of the ChineseAcademy of Sciences.中國科學院“百人計劃”研究項目資助
