
聚芴類半導體光譜穩定性
2010-11-30 10:57:00解令海石乃恩
物理化學學報 2010年4期
梁 婧 錢 妍 解令海,* 石乃恩
陳淑芬1,2 鄧先宇1,2 黃 維1,2,*
(1江蘇省有機電子與信息顯示——省部共建國家重點實驗室培育基地,南京 210046; 2南京郵電大學信息材料與納米技術研究院,南京 210046)
聚芴類半導體光譜穩定性
梁 婧1,2錢 妍1,2解令海1,2,*石乃恩1,2
陳淑芬1,2鄧先宇1,2黃 維1,2,*
(1江蘇省有機電子與信息顯示——省部共建國家重點實驗室培育基地,南京 210046;2南京郵電大學信息材料與納米技術研究院,南京 210046)
有機半導體的物理和化學性質直接影響其光電器件的性能,這為物理化學提出了新的研究內容與挑戰.其中,聚芴類藍光二極管的光譜穩定性及低能發射帶(LEEB)的起源問題是國際上近十年的熱點問題之一.本文系統概述了低能發射帶的現象、表征方法以及可能的形成機理,包括鏈間作用導致的激基締合物發射、器件制備或降解過程形成的芴酮缺陷發射、芴酮聚集態發射以及聚芴端羥基界面氧化導致的綠光發射.本文綜述各種物理摻雜和界面調控改善聚芴類二極管藍光穩定性的策略,著重論述非平面基團的空間位阻、分子構象與鏈的拓撲結構以及引入抗氧化受阻胺光穩定劑來提高其光譜穩定性策略.
電致發光;共軛聚合物;聚芴;低能發射帶;光譜穩定性;藍光半導體
塑料電子的新熱潮始于1990年,英國劍橋大學Cavendish實驗室Friend等[1]首次報道基于聚(苯乙烯撐)(PPV)半導體的高分子發光二極管(PLEDs). 1991年,Ohmori等[2]首次報道了聚芴應用于PLEDs.自此,聚芴作為寬帶隙半導體(Eg≈3.2 eV)因其高的固態量子效率、良好的載流子傳輸能力和較好的熱穩定性等優點,已經成為最具有商業潛力的聚合物藍光半導體材料之一,并受到塑料電子學領域廣泛關注.最近,大量綜述總結了其合成方法、結構-性能關系以及器件應用等方面[3-11],如Springer出版了《Polyfluorene》[12].然而,全面深入理解聚芴光譜穩定性綜述報道仍很少,特別是低能發射帶(LEEB)現象(部分文獻稱為long-wavelength emission),它直接影響器件的壽命及伏安特性(I-V)、發光效率、色純度等性能,涉及器件結構中的缺陷與界面等物理與化學過程,對聚芴類半導體的PLED顯示與照明器件中的應用與其商業化進程至關重要,成為繼聚合物半導體分子能帶工程的又一重要研究課題.然而,聚芴類半導體的光譜穩定性涉及電化學、熱化學、表面形態以及環境影響等方面.在本文中,我們組將對聚芴光譜穩定性LEEB現象進行文獻綜述,并闡述各種表征方法在論證LEEB現象的起源方面的研究進展;討論由于化學結構光或熱降解、氧協同的氧化作用、器件結構界面的降解、分子間作用導致形態演化等多種LEEB形成機制,系統全面概述其穩定化策略,最后,對該領域進行總結并展望.

1996年Pei等[13]制備了基于聚芴衍生物(BDOHPF)1的高效藍光薄膜(量子效率達73%),應用于PLEDs和發光電化學電池(LECs),并觀察到薄膜的LEEB現象.圖1是其薄膜態的光致發光(PL)λ=367 nm,主發射峰為430和450 nm,同時伴隨兩個肩峰480和520 nm.1998年Miller等[14]報道了(DHF-15-DPF)2電致光譜中的520 nm的LEEB現象(圖2),并將其歸因于成膜狀態下聚芴鏈間聚集作用的結果.在其隨后的研究中,關于聚芴類半導體的LEEB現象的機理以及起源成為該領域爭論的熱點問題[5],同時,多元化分子工程光譜穩定化策略相繼被報道.

圖1 BDOH-PF在溶液和薄膜態的發射光譜[13]Fig.1 PL spectra of BDOH-PF in solution and film[13]
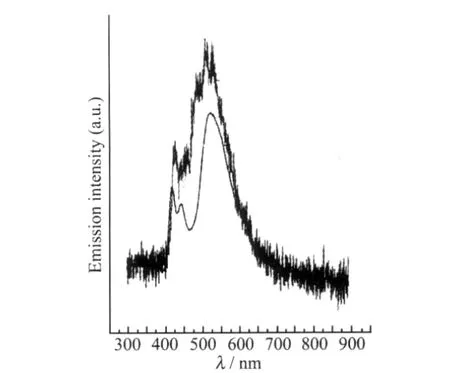
圖2 DHF-DPF的電致光譜與200℃熱退火1 h的熒光光譜[14]Fig.2 EL spectra and PL spectra of DHF-DPF after annealing at 200℃for 1 h[14]
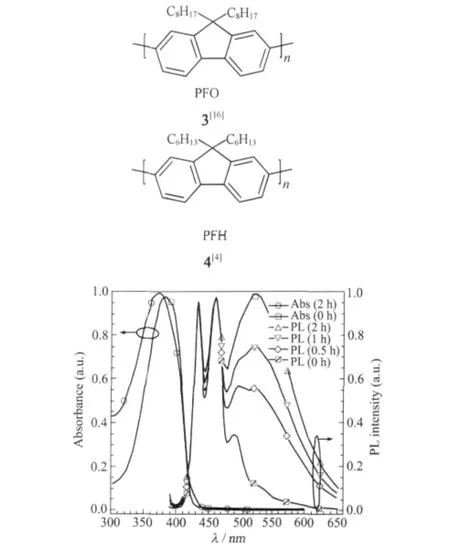
圖3 空氣中PFO不同紫外光照時間前后的紫外、熒光光譜[16]Fig.3 Time-dependent UV and PL spectra of PFO in air before and after UV irradiation[16]
聚芴類半導體的LEEB現象主要體現于溶液態或者薄膜態的PL光譜行為和EL光譜行為,通常對芴類材料藍光穩定性研究的監測方式包括在100℃以上高溫退火[15],常溫紫外光照[16]以及電致發光(EL)器件運行過程的測試[17].下文中將聚(9,9-二辛基芴)簡稱(PFO)3[16],聚(9,9-二己基芴)簡稱(PFH)4.圖3是PFO薄膜在空氣中不同光照時間的紫外吸收(UV)和熒光(PL)光譜圖[16],隨輻射時間增加,綠光的LEEB強度明顯增加,導致色坐標漂移和色純度下降.據目前研究來看在多數情況下,LEEB現象在多數情況下是指在520-530 nm出現的綠光帶(又稱gband)[17].然而,據文獻報道,LEEB現象還有其他多種表現:(1)2000年Weinfurtner等[18]研究了PFO分子量和聚集行為及PLED光譜穩定性的關系,報道了510 nm為中心的LEEB現象(器件結構為ITO/PEDOT/ PDOFO/Ca,圖4).(2)Chen等[19-20]觀察到通過XRD表征證實了結晶聚芴的端基相互作用,并指認了507 nm的LEEB來源于未封端聚芴鏈的端基聚集(圖5);(3)電場導致的交聯結構可以加強聚集從而產生LEEB現象[21],其LEEB主峰位于485 nm,有明顯的精細結構,位置和形狀都不同于沒有精細結構的520-530 nm的綠光峰.

圖4 PFO的熒光光譜及隨時間變化的電致光譜[18]Fig4 PL and time dependent EL spectra of PFO[18]
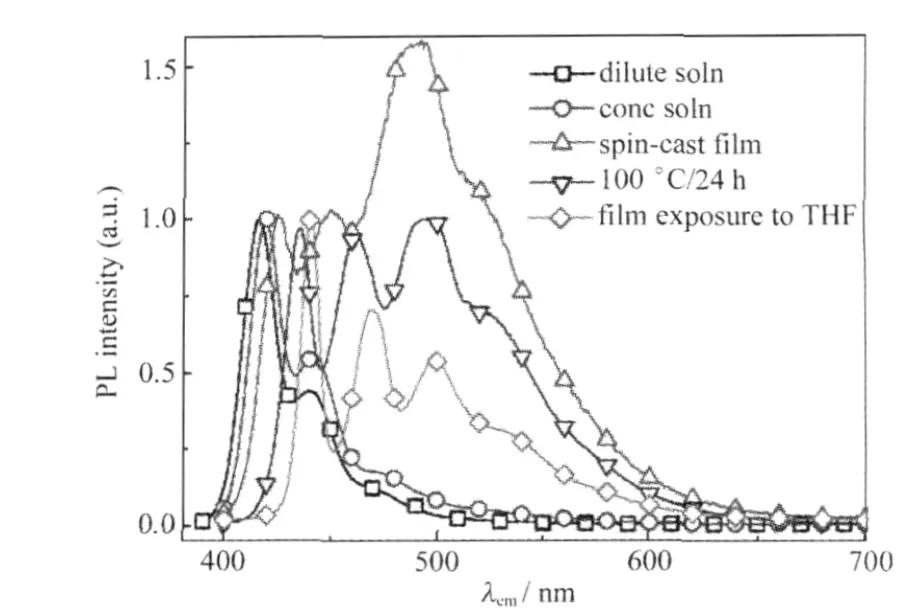
圖5 未封端的PFO在不同聚集態的熒光光譜[20]Fig.5 PL spectra of uncapped PFO under different aggregation conditions[20]
具有特殊聚集態結構的PFO薄膜表現出不同光致發光行為.Su等[22]系統制備了各種PFO的相結構,包括N相、β相、α相、α′相,通過X射線衍射和電子顯微鏡進行表征[23],同時研究了在380 nm激發光下薄膜光譜行為(圖6).其四種相的光譜都表現為三組發射帶,其中PFO β相的0-0躍遷帶在438 nm處,相對N相有11 nm紅移.據研究表明,這歸因于β相中PFO特殊的構象與聚集行為,β相中鏈內芴-芴扭曲角接近平面的180°會導致其共軛鏈長延長.
定量研究LEEB現象為對比與篩選材料提供了重要依據,White[24]和 Huang[25]等利用綠光指數(green index)定量描述了LEEB現象,其定義是聚芴的發射光譜500 nm以上綠光峰與500 nm以下藍光峰的強度之比,圖7為修飾有POSS位阻基團的PFO膜(PFO-POSS)5[24]在空氣中200℃退火的LEEB光譜,其綠光指數由退火前的0.05提高至3.70.

圖6 不同相結構的PFO發射光譜[22]Fig.6 PL spectra of PFO with different phases[22] The normalized spectra were given as inset.

圖7 PFO-POSS膜LEEB光譜[24]Fig.7 LEEB spectra of PFO-POSS film[24]

目前文獻報道了大量的LEEB現象,通過各種儀器表征手段試圖證明其機理.迄今,LEEB可能的形成機制主要包括:分子聚集及其激基締合物(excimer)發射、光氧化、熱氧化、電氧化(器件運行過程)以及材料合成中導致的芴酮結構的缺陷等.但是由于聚芴及其衍生物材料的種類不同以及實驗條件的差異,這些對深入研究LEEB現象的起源與衡量材料穩定性帶來很大困難.迄今,LEEB的起源仍困擾著材料學家:早期的研究者支持聚集或excimer機理,只是通過對比稀溶液和濃溶液,溶液態與薄膜態來定性研究聚集體,沒有定量的證據[26];然后,基于芴酮缺陷觀點基本確立并占據主流,Scherf等[27]堅持LEEB(530 nm)主要來源于鏈上芴酮缺陷;相當多的文獻中刻意將芴酮單體通過共聚引入聚芴,得到了與正常降解過程極為類似的綠光發射帶,進一步確認單鏈芴酮是LEEB的可能發色團[17].最近,聚集機理的研究又有了新的報道,高純的三聯寡聚芴在不良溶劑中聚集產生LEEB的實驗有力支持該觀點[28].另外,聚集機理也可以從早期研究中通過共聚扭曲基團、9-位取代樹枝化等化學改性途徑可以減少LEEB的事實中找到依據.盡管材料改性方面較多可以通過抑制鏈間聚集來進行分子設計[17,29],但是支持聚集機理的研究者為數不多.
1 LEEB形成機理
Bliznyuk研究組[30]最早對LEEB的起因做了系統的研究和闡述,他們在聚芴藍光材料及器件的光降解和電降解過程中觀察到LEEB,將LEEB來源歸屬于兩個過程:光化學氧化過程以及物理聚集態變化過程,前者通過氧化產生了芳基酮,猝滅發光;后者在形態衍化中發生π發色團堆積形成了激子缺陷,減弱發光強度并使發光帶紅移.
1.1 聚集或激基締合物機理
Bradley研究組[31]的早期研究表明在非理想溶劑或器件降解中,聚合物易發生聚集,可以通過紫外光譜觀察到433 nm的聚集特征吸收峰.Miller等[14,32-33]系統研究了PFO的薄膜態和溶液態的吸收和發射光譜差異,發現二者吸收光譜近似一致,在熱處理實驗中,僅觀察到分子鏈排列較為緊密的薄膜聚集態的長波發射光譜,據此推斷出LEEB來自鏈間相互作用導致的激基締合物(excimer)發射.對聚集機理的研究進行總結,影響聚芴熒光光譜的聚集體主要包括:聚芴主鏈聚集體、聚芴鏈的端基聚集體和聚芴鏈間交聯結構三種類型,以下將從熱處理和光處理兩方面討論聚集對LEEB的貢獻.
我們組[34]對聚芴及其衍生物熱穩定性的早期研究支持和證明了聚芴聚集體在產生綠光發射過程中的主要作用,實驗對比150℃下空氣和氮氣退火后聚芴薄膜的發射光譜,都觀察到相近的520 nm發射峰,排除了氧氣氧化產生芴酮結構對LEEB的主要貢獻;將空氣中200℃退火后的薄膜,用干冰-甲醇浴淬火,并分散聚合物分子鏈,此時出現在發射光譜中的LEEB消失,證明聚集產生的分子間激基締合物是LEEB發射的主要原因.
為研究端基聚集與LEEB之間的關系,Chen等[20]研究分別有封端與無封端的聚芴在100℃退火24 h前后的光譜變化,以苯封端的聚芴只有藍光發射,而端基為溴的未封端樣品觀測到明顯的聚集體的特征吸收帶(431 nm)及相應的綠光發射峰(507 nm),且傅里葉紅外光譜(FTIR)檢測無1721 cm-1的酮式特征吸收,因此推測其來自端基的聚集.另外, Chen等[35]還設計了有鏈間相互作用的聚芴分子(POBOHF)6,測試其在不同電壓產生綠光帶的情況(器件結構為ITO/PEDOT/POBOHF/Ca/Al),并與PL峰對比,發現其電致發光光譜在5 V電壓下,第一次掃描即出現424、452和485 nm的寬發射帶,并且有520和570 nm的肩峰(圖8).隨掃描次數增加和掃描電壓的增大,藍光峰下降和綠光峰上升的程度均增加,且在掃描電壓重復第6次時,420-460 nm藍光峰幾乎消失,由485-550 nm綠光峰代替,說明綠光帶來自電場導致的鏈間聚集相互作用.最近,對POSS大基團封端的聚芴的研究表明熱處理后伴隨的交聯反應對產生LEEB有重要作用,他們認為可能是由于其阻止了鏈扭曲,增大了能量躍遷到缺陷上的幾率從而導致明顯的綠光發射[24].
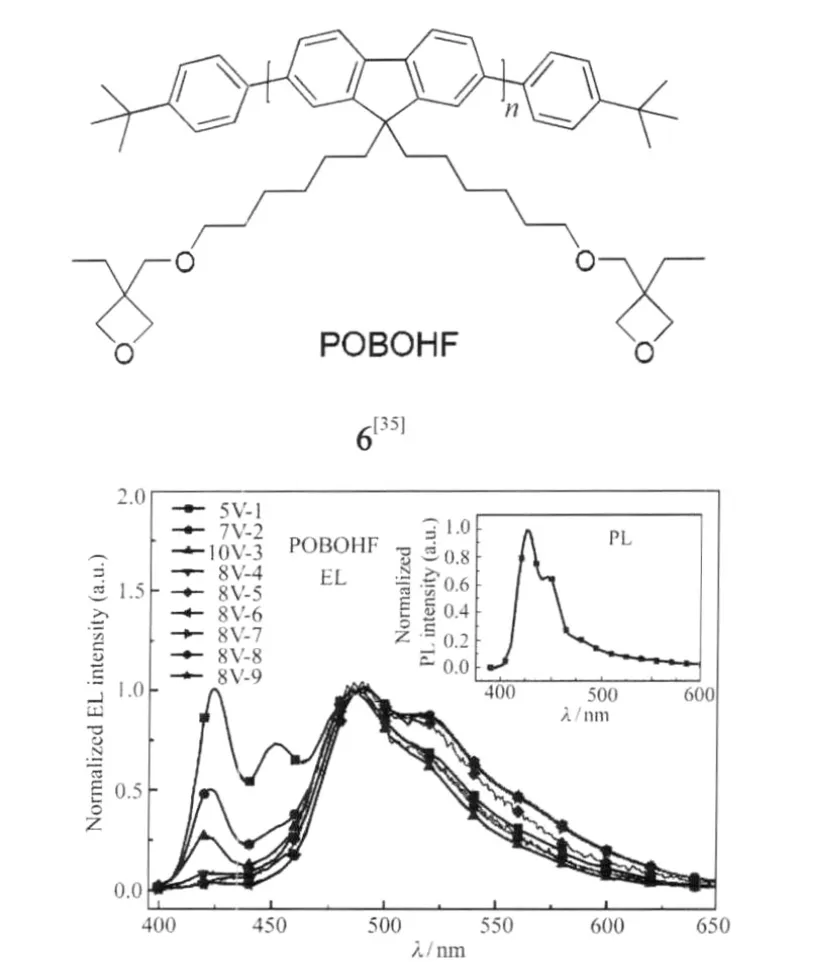
圖8 POBOHF電致與光致發光光譜[35]Fig.8 EL and PL spectra of POBOHF-based device[35]
為了說明聚芴鏈間堆積得到綠光發射的原理, Chen等[36]設計了側鏈修飾帶有咔唑基團烷基鏈的(Cz100PF)7,測試PFO和Cz100PF的紫外吸收、光致、電致發光光譜(圖9),并借助時間分辨的電致發光光譜儀(time-resolved(TR)EL)以及分子模擬軟件,將Cz100PF的藍光發射歸屬于聚芴的主鏈發射, 530 nm的綠光發射帶歸屬于咔唑側鏈之間堆積形成的雙分子聚集體導致的電致復合物(electroplex).

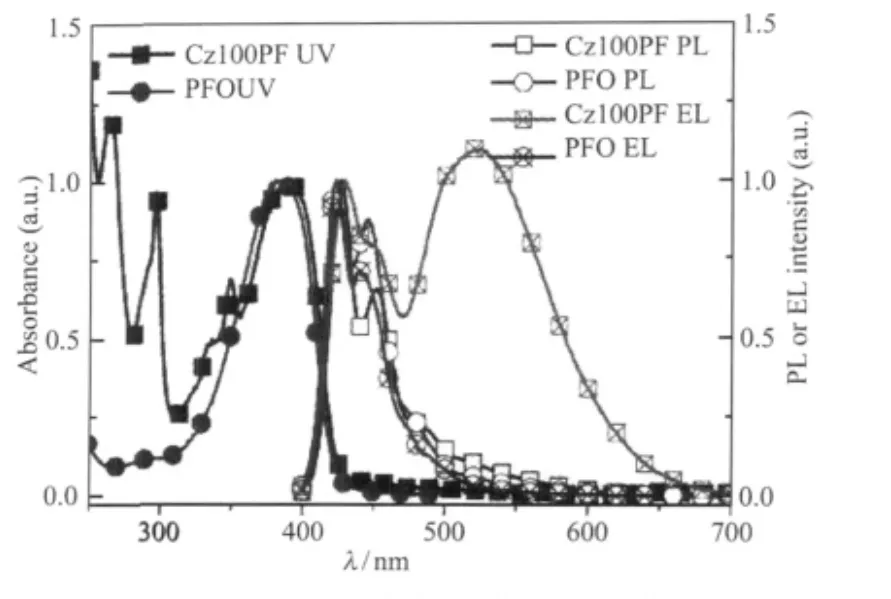
圖9 PFO和Cz100PF的紫外吸收、光致、電致發光光譜[36]Fig.9 UV,PL and EL spectra of PFO and Cz100PF[36]
溶液聚集態研究為聚集導致的綠光帶提供更有力的證據,早期研究分子間氫鍵作用的聚芴衍生物Polymer 1(簡稱P1)8[37]溶液的穩態和瞬態熒光光譜(圖10),當減弱P1分子間的氫鍵聚集作用時,其光譜形態由非指數衰減變為單指數衰減,預示聚集作用的減弱,且LEEB減弱并消失,表明觀察到P1在聚集態形成的激基締合物發射與LEEB的對應關系,而不存在分子間氫鍵作用的Polymer 2分子無此現象;在穩態熒光光譜表征中,具有分子間氫鍵作用的聚芴薄膜態表現出明顯的聚集綠光發射帶,而無分子間作用的聚芴則沒有此發射峰.最近, Koizumi等[28]設計比較了親油性(DHF3)和兩親性(Am-F3)9三聯寡聚芴在THF溶劑或THF/H2O混合溶劑中的光物理行為(圖11),發現Am-F3在THF/H2O混合溶劑中發生明顯的分子凝聚現象,在540 nm處出現了尖銳的綠光帶發射,由于合成的寡聚芴的純度相當高,通過結構表征排除了芴酮的存在,而后將聚集體繼續溶解在THF溶劑中,綠光帶消失而藍光發射重新出現,這說明寡聚芴單元的聚集對綠光發射帶的產生起了關鍵的作用.

圖10 (a)紫外吸收光譜與光致發光光譜,(b)四氫呋喃溶劑和四氫呋喃-甲醇溶劑中的時間分辨熒光光譜[37]Fig.10 (a)Abs spectra,PL spectra,(b)time-resolved photoluminescence decays for polymer in THF solution and THF/methanol solution[37]
最近,rod-coil分子也被用來研究聚芴光穩定性機理[38-40].為了辨別究竟是聚集發射還是芴酮發射在聚芴光降解現象中起主導作用,Chochos等[40]設計了含有不同位阻長度的寡聚芴單元的剛柔嵌段(rodcoil)結構的模型分子,研究其光處理后的發射光譜,得到了只要合理控制鏈間距離,即使有芴酮結構,也沒有觀察到綠光帶的結果,有力地證明了聚集作用而非芴酮因素是綠光發射的主導因素.
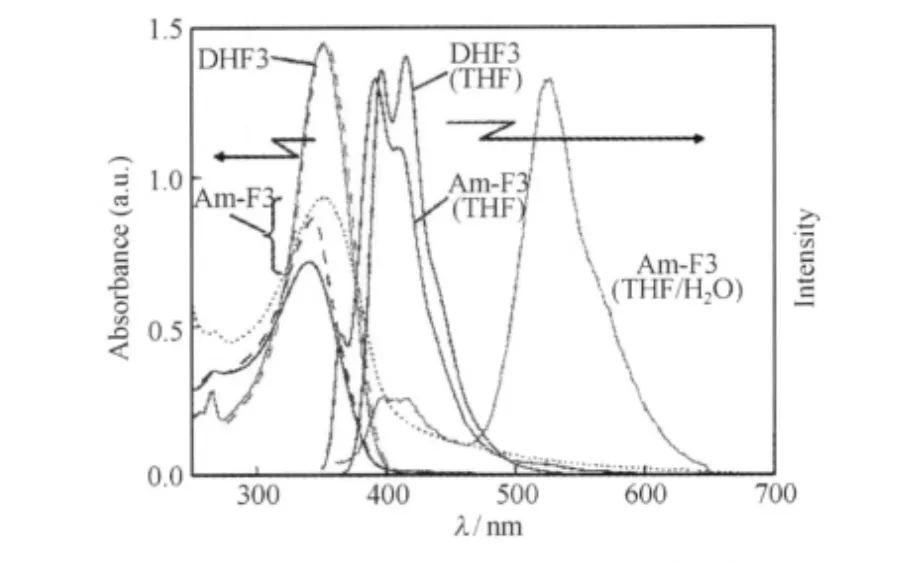
圖11 Am-F3和DHF3在四氫呋喃-水溶液中的吸收和發射光譜[28]Fig.11 Abs and PL spectra of Am-F3 and DHF3 in mixtures of THF/H2O[28]
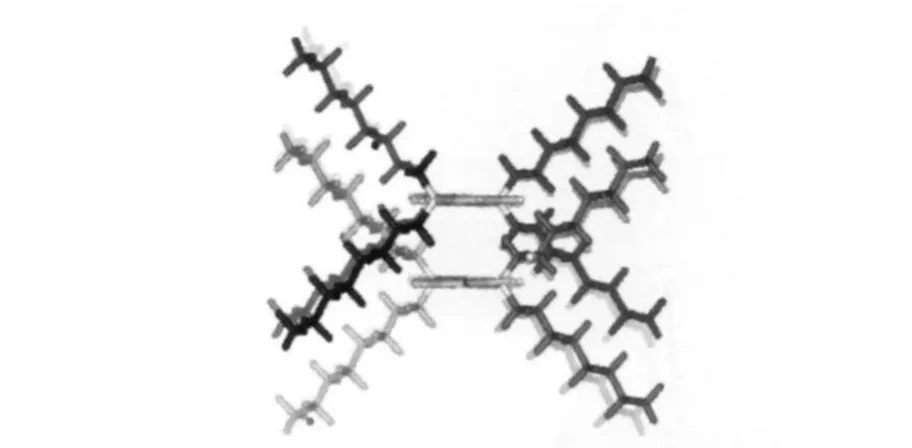
圖12 PFO的穩定雙鏈堆積模型[41]Fig.12 Stable packing modes of two chains of PFO[41]
分子模擬研究是研究共軛聚合物鏈相互作用的理論方法之一.針對PFO體系的薄膜聚集態的分子動力學模擬[41]也有力地支持了上述結論(圖12),且指出有效堆積的必要條件為具有合適長度和取代基的基團堆疊[42],且發生鏈間π軌道的有效堆積可以產生激基締合物長波發射.
1.2 芴酮缺陷機理
List等[43]提出LEEB起源于鏈上的酮式缺陷.通過廣泛研究聚芴的降解機理和535 nm LEEB的來源,他們認為芴酮結構相當于聚芴鏈上低能量的發光客體或者激子陷阱,發生由聚芴向芴酮的F?rster能量轉移,使激子傳遞中被捕獲陷落,產生猝滅藍光和LEEB現象.表征中,通過傅里葉紅外光譜(FTIR)檢測到空氣中光降解后,聚芴出現在1721 cm-1的羰基特征吸收峰(圖13)[43];且該吸收峰強弱(即芴酮濃度)與綠光發射強度有關[44-45],另外,ESCA譜中觀測到O的信號也佐證了芴酮的存在[30].
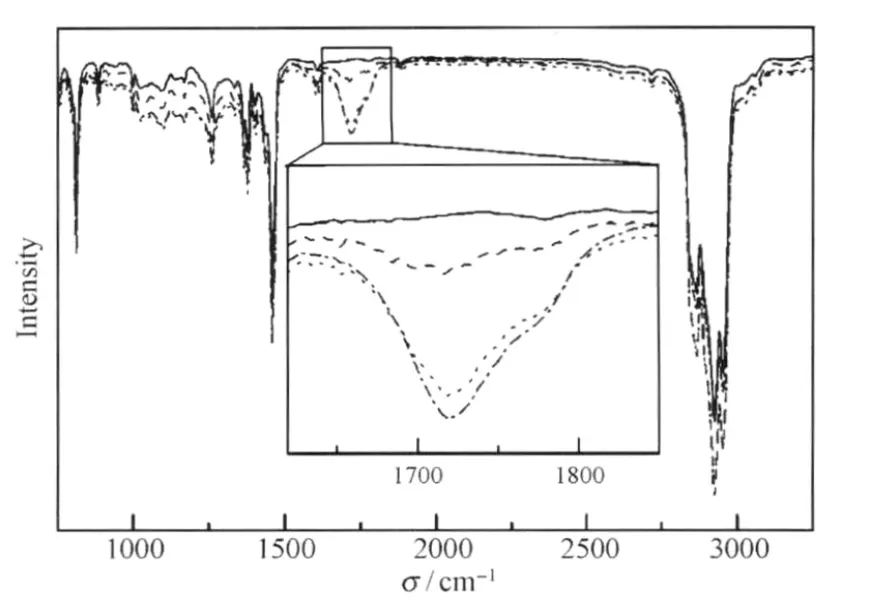
圖13 PFO中芴酮結構的FTIR譜[43]Fig.13 FTIR spectra of fluorenone defect in PFO[43]
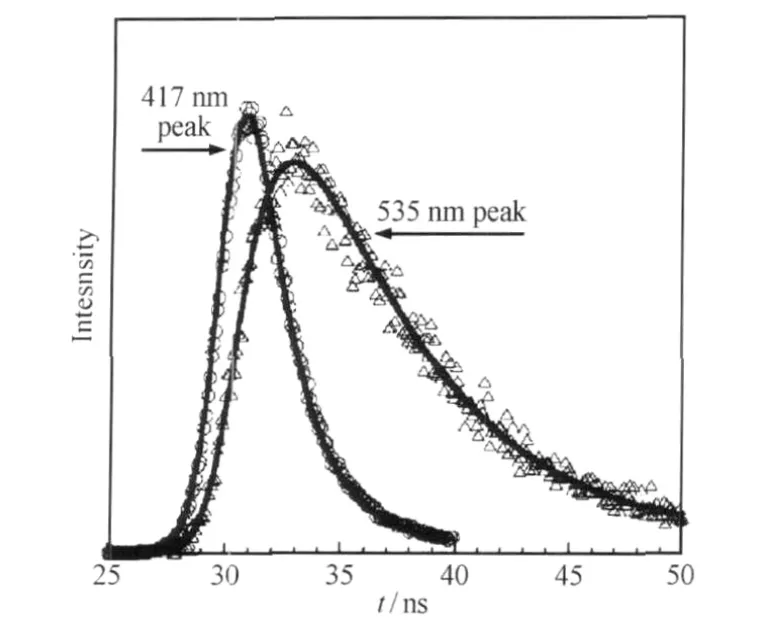
圖14 PFN分別在417與535 nm檢測波長下的瞬態熒光光譜[48]Fig.14 Fluorescence decay emission of PFN detected respectively at 417 and 535 nm[48]
在闡述芴酮導致LEEB的過程中,材料學家通過不同的方法,從理論計算、合成特定的模型分子,到研究含芴酮的聚芴或無缺陷的聚芴的不同的熒光壽命,都說明了LEEB來源于不同途徑產生的芴酮缺陷.Zojer[46]和Franco[47]等引入量子化學計算的方法,前者證實強的能量轉移和芴酮激子的強定域作用使聚芴上含有0.1%-2.0%的芴酮單元就足以產生非常明顯的LEEB;后者認為芴酮不是激子猝滅基團,而是作為純的聚芴材料的黃色發光客體,由于摻雜產生LEEB發射.Jenekhe等[48]以含有不同比例芴酮基團的共聚芴(簡稱PFN)為模型分子,研究其光譜行為,發現含3%芴酮的共聚芴的溶液在10-7mol·L-1的濃度下就有明顯的535 nm綠光帶,圖14為瞬態熒光光譜,PFN在10-6mol·L-1甲苯溶液, 381 nm紫外光激發下,其535 nm的熒光壽命在5-6 ns,呈現單指數衰減,大大超出420 nm藍光發射帶的壽命(約240-400 ps),暗示芴酮結構是綠光發射的唯一發色基團,且作者將聚集導致的綠光強度的增加解釋為鏈間相互作用增加了從芴到芴酮基團的能量轉移加強.
為了分辨LEEB究竟是來自芴的還是芴酮羰基的n-π*躍遷,Wegner等[49]通過瞬態熒光光譜得到了共聚芴酮的寡聚芴(OF5K)10分別在365 nm(對應π-π*躍遷)和453 nm(對應n-π*躍遷)的光激發下的熒光壽命(圖15),兩種激發波長下,長波發射熒光壽命都在8 ns左右,表明LEEB更可能來自芴酮的發射.

圖15 OF5K的熒光強度對時間依賴關系[49]Fig.15 The time dependence of PL intensity for OF5K[49]
Ma等[50-51]最新提出的支鏈芴酮機理是對經典芴酮機理體系的補充,研究表明芴9-位取代烷基鏈上的酮結構也對LEEB發射和藍光猝滅有重要貢獻,提出PFO可能的降解產物11.實驗中,他們合成了聚芴、烷基酮、芴酮結構單體,進行不同濃度的共混,研究其對綠光帶的非輻射猝滅作用,發現綠光帶強度與烷基酮濃度有依賴關系:在烷基酮存在的條件下,極低濃度的芴酮即能引起低能綠光發射.
對于芴酮缺陷的來源爭論中,金屬催化劑的有意或偶然引入對芴酮的形成有重要的作用.List等[43]研究包括在Yamamoto聚合反應中Ni(0)誘導的單烷基鏈氧化而原位生成的芴酮缺陷和由于光和熱氧化導致雙烷基鏈降解而形成的芴酮缺陷,認為Ni(0)在芴酮缺陷形成中起到觸發劑的作用.而有人[16,35]認為器件中界面效應導致Ag陽極中含有的Ca離子可能對形成芴酮結構有貢獻.Ma等[52]認為偶聯聚合的催化劑Pd(0)對生成芴酮有促進作用. Holmes等[53]的研究表明,單體合成中的單烷基缺陷在芴酮的形成過程中扮演重要角色,只要能將其避免,無論用Ni(0)催化的Yamamoto聚合或是Pd(0)催化的Suzuki聚合得到的聚芴均不產生LEEB.然而,Suranna等[54]研究四聯寡聚芴的光譜穩定性后提出了不論是單烷基芴還是二烷基芴都可能經歷自由基降解過程而形成了芴酮,其可能的降解過程如圖16所示.他們發現單烷基芴并非芴酮的唯一來源,這可能只是自由基降解的開端,而添加了Ni(0)金屬催化劑雜質后卻明顯觀察到了綠光帶的發射,Ni的觸發氧化功能再次被證實.
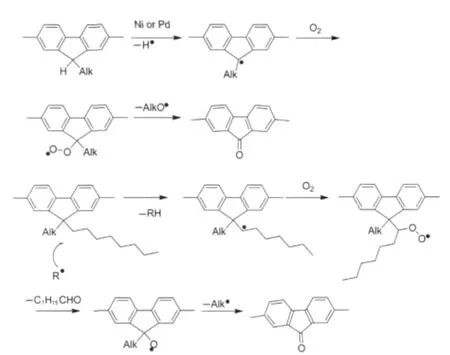
圖16 氧化生成芴酮缺陷的自由基氧化途徑[54]Fig.16 Pathways of fluorenone defect formation with radical oxidation[54]
最近,Meijer等[55]提出,芴酮的產生與聚芴9-位的取代側鏈有重要的聯系,他們通過循環凝膠滲透色譜法(rec-GPC),以及基質輔助的激光解吸電離飛行時間質譜法(MALDI-TOF MS)對聚芴的降解過程進行表征(圖17),在GPC譜上觀察到因為氧化產生的相對分子質量高于原始峰的兩個新峰,MS表征得到增加了一個或兩個氧原子的質譜峰數據,據此推斷,聚芴的降解過程可能包含羧基化或者酮式中間體,其可能的降解過程如圖18所示.

圖17 五聯寡聚芴在空氣中降解后的凝膠滲透色譜和質譜[55]Fig.17 GPC and MALDI-TOF MS spectra of pentafluorene after degradation in air atmosphere[55]

圖18 寡聚芴的光降解和熱降解機理[54]Fig.18 Proposed mechanism for pentafluorene residue by irradiation in air and thermal treatment[54]
Lu等[56]獲得了環丙基螺芴(BFSCF)12的單晶堆積結構(圖19),螺芴分子成十字型排列,分子間距離為0.27-0.33 nm.晶體結構中沒有出現通常意義上聚芴鏈間π-π面面堆積作用,主要表現為CH…π相互作用.當在空氣中150℃熱退火時,模型化合物產生了明顯的520 nm綠光帶,因此可以排除芴鏈聚集機理,說明LEEB主要來自材料或器件制備過程中殘余氧影響下產生的芴酮發射.

圖19 BFSCF的雙分子堆積的單晶結構[56]Fig.19 Single crystals of double molecular stacking for BFSCF[56]
最近,Ma等[57]對三聚芴13的研究表明,其分子間面面堆積作用的距離為0.395 nm(圖20),在此距離下難以發生π發色團的相互作用.為證明LEEB來自芴酮單體發射,而非聚集體發射,Ma等[58]采用9-位單芳基取代或芳基和甲基取代的梯形聚芴14[58],對比研究純凈的和降解后(200℃下空氣中退火2 h)MeLPF的甲苯溶液(50 μg·mL-1),于室溫和77 K時400 nm激發下的PL光譜變化,證明有氧氣參與的熱降解可以產生芴酮結構.通過惰性分散介質將降解的HLPF鏈間距分散至12 nm,已經超過了鏈間堆積相互作用的距離(0.4 nm以下),依然有較強的LEEB發射,闡明LEEB僅來自降解產生的酮式發色團.

1.3 芴酮激基締合物機理
與聚芴鏈間聚集以及芴酮單分子發射機理不同,芴酮聚集體機理主要指出鏈間或者鏈內的芴酮單元之間相互作用形成的激基締合物或者激基復合物導致的LEEB現象.
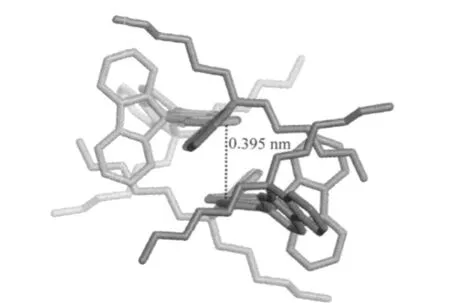
圖20 三聚芴單晶的π堆積結構[57]Fig.20 Molecular π-stacking diagram of fluorene trimer[57]
Bradley等[17]率先提出鏈間芴酮聚集體產生的非定域的激基締合物(fluorone-based excimer)導致了LEEB的形成,其實驗設計將PFO分散到聚苯乙烯聚合物陣列中,在光氧化后檢測到芴酮缺陷,卻沒有觀察到明顯的綠光發射現象,對比實驗中,沒有分散的PFO在同樣條件處理后卻產生了明顯的LEEB發射,他們歸因為聚苯乙烯分散了PFO由于降解產生的芴酮缺陷而無法形成鏈間的芴酮聚集體或激基締合物,因而提出LEEB來自非定域的芴酮激基締合物發射.隨后,Chochos等[40]對在聚苯乙烯中分散的rod-coil嵌段三聚芴進行光降解處理,也觀察到類似的實驗現象,并且觀察到420 nm左右藍光發射的猝滅和LEEB現象并不是同時出現,說明前者可能由于鏈上由芴到芴酮的能量轉移導致,后者由于鏈間芴酮結構的聚集導致.
Bo等[59]研究表明鏈間的芴酮相互作用產生并增強了LEEB發射,采用時間分辨熒光光譜研究了PFN與不同迭代長度的樹枝狀位阻的共聚芴的共混薄膜,表明樹枝狀基團可以顯著分散PFN鏈,并減少鏈間芴酮的聚集體導致的LEEB現象.
最近,Bradley等[60]的研究進一步表明:LEEB來自鏈間芴酮結構的面面堆積形成的激基締合物態.他們觀察到聚苯乙烯分散的PFO在高于玻璃化轉變溫度的熱退火實驗中也產生了LEEB現象,說明熱降解改變了聚合物的鏈間規則度,增強了鏈間相互作用.另外,當將綠光發射聚合物重新溶解或者重新涂膜,LEEB現象消失(圖21),證明鏈間聚集體為LEEB的必要條件.

圖21 降解產生綠光帶的PFO薄膜光譜及其溶解后重新成膜的光譜[60]Fig.21 Photoluminescence spectra of degraded PFO film with LEEB and it re-casting film[60]

圖22 不同極性PFN(10-5mol·L-1)的LEEB光譜[61]Fig.22 LEEB spectra of PFN solution (10-5mol·L-1)of different polarity[61]
Pei等[61]研究比較了共聚不同比例芴酮單體的PFN的穩態熒光光譜和時間分辨熒光光譜,指出LEEB來自鏈內的芴酮單元間的相互作用.圖22為芴酮濃度為12.5%的PFN分別在不同極性溶液中的穩態熒光光譜,當在良溶劑氯仿中,PFN呈單分散態,LEEB幾乎觀察不到,隨著溶液極性的變化, PFN的鏈扭曲作用增強,產生了明顯的LEEB發射.另外,對時間分辨光譜的研究表明,溶液態的單分散PFN產生的LEEB熒光壽命最長,在固態薄膜中,當PFN中芴酮單體濃度從0.1%至25%變化時,其LEEB熒光壽命從8.15 ns減小至4.21 ns,均小于鏈上芴酮單體的熒光壽命8.39 ns,說明了鏈內的芴酮基團的相互作用起主要作用.
在綠光究竟來自芴酮單鏈還是芴酮鏈間或者鏈內聚集體的爭論中,值得一提的是,Lupton等[62-63]堅持LEEB來自單鏈芴酮結構的發射,研究含有兩個面面堆積芴酮結構的模型分子15,沒有發現分子內的締合物發射信號,但是芴酮之間有較強的發光猝滅作用,用單分子發射光譜檢測法研究PFO的光譜性質,發現綠光發射的強度與芴酮的含量有較強的依賴關系.同時,Wegner等[49]通過單晶堆積結構(圖23)證明含有芴酮的聚芴鏈的鏈間堆積作用不可能形成芴酮的激基締合物,因此聚芴綠光帶只能來源于9-位芴酮的單分子發射,而非芴酮聚集體激基締合物的發射.

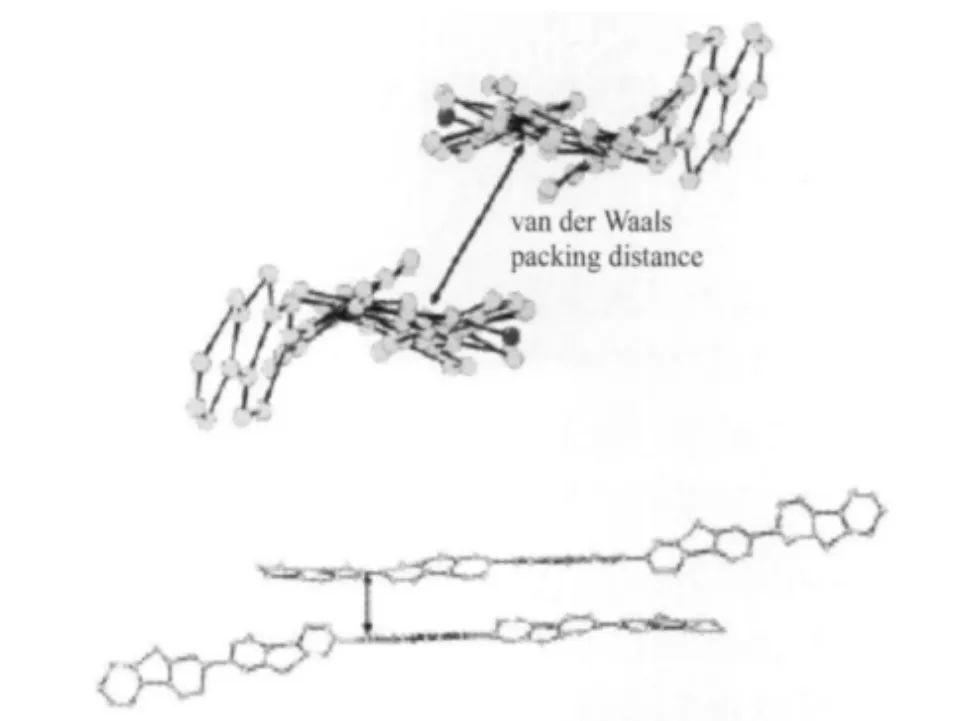
圖23 OF5K的分子堆積示意圖[49]Fig.23 Molecular packing modes of OF5K[49]
1.4 聚芴端基界面氧化機理
作為對經典聚集以及芴酮機理的補充,Kappaun等[64]的最新研究表明,在解釋界面缺陷對LEEB形成的貢獻上,LEEB的產生可能源于端羥基在Ca2+離子催化下氧化去質子過程生成氧負離子基(圖24).與傳統的綠光帶位置不同,此低能發射帶主要位于500 nm.此前,Moses[16]和Chen[35]等認為電致發光器件中,Ag陽極中含有的Ca2+離子可能催化PFO發光層氧化產生了酮式結構,導致發光的猝滅和LEEB現象的產生;然而聚芴端基的界面氧化機理很好闡釋在PLED器件中,金屬Ca或Ag電極與聚芴發光層之間相互作用的實質.
總結上述四種機理,我們認為形成綠光帶的原因復雜,芴酮和聚集可能都是綠光發射帶的主要產生原因,二者在不同的條件下各占主導作用.首先,綠光發射與聚集態關系密切,聚芴薄膜態與溶液態相比更易觀察到綠光發射,而溶液態中即使有少量由于降解產生的芴酮單元,也因為分散鏈減弱能量轉移而使綠光發射并不顯著;其次,無論是芴酮的激基締合物還是芴酮單鏈而導致LEEB,都說明芴酮是一個不可忽視的要素,其中兩點已得到公認,一是存在由聚芴主鏈上的單重激發態到芴酮單元之間存在能量轉移;二是芴酮缺陷可以捕獲載流子,猝滅發光.

圖24 含硼酸端基的聚芴的降解機理示意圖[64]Fig.24 Degradation mechanism of polyfluorene with boric end groups[64]
然而,前期對聚集作用的研究幾乎都只是定性表征,缺乏對聚集產生綠光發射的臨界鏈間距離的確切證據.Ma等[58]最近實驗可以更好闡明聚集態和芴酮發射的相關性:當在聚碳酸酯中分散不同濃度的降解(d-HLPF),在其濃度為0.1%-5%時,都沒有明顯的LEEB,濃度增加為10%,LEEB顯著,此狀態下鏈間距約1.2 nm,遠大于鏈間發色團堆積形成excimer的距離,但是能夠發生有效的F?rster鏈間能量轉移,說明由聚集導致的聚芴鏈到芴酮鏈上的能量轉移是LEEB的必要條件.
設計材料的策略中,我們發現控制聚芴主鏈的排列,保持適當的鏈間距至關重要,即使有芴酮產生,也能通過避免聚集導致的鏈間能量轉移來消除綠光發射;然而,鏈間距離不能無限增大,π共軛聚合物分子間是通過共軛鏈間的堆積作用傳遞載流子,超過臨界距離會由于影響載流子傳遞而降低器件效率.再者,總結芴酮的形成原因,從聚合過程中金屬催化單烷基芴的氧化,到ITO釋放的氧和金屬電極催化氧化,再到熱光電誘導的氧化過程等,都可以看出這是一個多元化的過程.因此,純化材料和選擇合理有效的器件結構也是提高材料壽命的一條重要途徑.
2 聚芴光譜穩定化策略
近年來,在研究LEEB起源問題的同時,科學家提出了多種穩定化策略,包括從共混摻雜到器件界面工程再到分子修飾.例如,Neher[65]和Jenekhe[66]等提出通過共混小分子的空穴傳輸材料或高玻璃化轉化溫度的聚合物的策略,雖然可以提高發光效率,但只能在一定程度上抑制LEEB的產生.下面將從抑制鏈間相互作用或芴酮缺陷的角度評述文獻報道的各類策略.
2.1 位阻與構象抑制鏈間相互作用
2.1.1 具有位阻效應的9-位碳3D樹枝或螺環化

Müllen等較早地將具有剛性的芳基位阻基團引入到芴9-位材料16[67],主要是利用屏蔽效應來阻隔剛性的聚芴分子鏈.Shu[68]和Carter[69]等通過不同的合成方法將三維的樹枝化結構引入芴的9-位,同時,為保持聚芴材料良好的溶解性和較大的聚合度,通常材料制備中使用的是與9-位烷基取代芴共聚的方法.樹枝化基團分為9-位直接與烷基相連或與芳基相連,二者相較:9-位直接連接烷基的聚芴較易于發生光氧化,而9-位直接與芳基相連,可以有效增大位阻和鏈間距,減少聚集.另外,有報道將具有空穴和電子傳輸性能的功能位阻結構[70-71]引入到芴的9-位,不僅提高了材料的熱穩定性,而且增加了材料與電極的匹配,能夠有效地提高發光效率.
芴9-位螺環化也是引入位阻基團的重要形式之一,主要包括引入環烷基、芳香基和雜芳基等結構.由于中心碳原子sp3雜化,使得螺結構的兩端構成非平面的空間結構,避免分子間聚集,從而達到穩定的藍光發射.
我們組最先將螺二芴引入聚芴體系17[72],發現螺芴結構使材料具有良好的熱穩定性和較高的玻璃化轉變溫度.同時由于螺芴大的空間位阻破壞了材料的結晶,降低了激子在鏈間的傳遞失活,抑制了激基復合物的形成,從而提高了材料的發光效率,得到較純凈的藍光發射;又通過增長螺位的長度,進一步改善了該類材料的載流子傳輸性能[73].Shu等[74]引入環烷基類18可以有效地提高溶解性,并能使材料具有良好的熱穩定性;以及通過螺環化將二苯乙烯結構引入聚芴主鏈上19[75],結果表明:在芴主鏈上光激發形成的激子能夠遷移到螺位,從而調制其發射波長.在最近的一些報道中,開始更多地重視通過在空氣環境中來檢測熱氧化以及光氧化對材料的降解情況的影響.其中,全螺結構的均聚芴20表現出良好的熱氧和光氧化穩定性[76-77].


最近,Ma等[78]合成了含咔唑基團的寡聚芴,外量子效率達3.72%,色坐標為(0.16,0.05),接近飽和的深藍光.另外,多螺環螺芴的合成研究發展迅速[79-83],器件數據表明其在電致藍光領域有很好的應用潛力.
2.1.2 共聚策略設計扭曲鏈構象抑制鏈間聚集
共聚引入功能基團,通常可以將9-位引入立體位阻基團、扭曲基團的芴單元與電荷傳輸基團或DA給受體基團等功能片段共聚,通過這種共聚方式優化共聚芴的功能性.Miller等[32]早期報道將蒽、聯苯基、半共軛的螺芴單元及苯基硅烷咔唑基團等作為扭曲結構團通過無規共聚引入聚芴體系21,有效地控制了共軛鏈長和抑制聚集,結果明顯地抑制了綠光發射.隨后,我們課題組[84-85]首次將可溶的烷氧基苯結構與芴單體共聚22,獲得了熒光效率為40%,最低啟動電壓為4 V的深藍光PLED器件.另外,將螺二芴作為扭曲基團引入聚芴也能得到高效的藍光材料23[86].

Wong[87]和Jenekhe[88]等設計的含有彎曲形骨架的共聚芴,前者含3,6-位咔唑等穩定結構24,后者引入2,3-二對亞苯基喹喔啉,通過提高其HOMO軌道能,明顯增強材料的熱穩定性和非平面性,減少聚集的產生.而Scherf等[89]將咔唑的9-位通過柔性的烷基連接后與芴共聚25,得到416 nm的深藍光,且經過200℃空氣退火后沒有出現任何長波發射,表明柔性鏈的引入成功地阻礙了芳環間的相互作用,其器件啟動電壓4 V,亮度400 cd·m-2.
共聚策略還可以通過引入相應的功能基團,增加材料與電極的匹配性.我們組[90]將芴單體與噁二唑取代苯共聚構建了主鏈為空穴傳輸通道,側鏈為電子傳輸通道的十字交叉型聚芴26,同時達到提高載流子傳輸性能和減弱鏈間相互作用的目的.另外, Liu[91]和Jen[92]等將四苯基硅烷引入共聚芴體系,通過共聚3,6-咔唑、二烷基芴或螺二芴得到了熱穩定性良好的寬帶隙藍光材料,如27[92],其電致發光波長位于409 nm處,器件結構ITO/PEDOT/polymer/ TPBI/CsF/Al,外量子效率2.4%,亮度850 cd·m-2,啟動電壓5 V.硅烷的引入降低了空穴的注入勢壘,有利于平衡載流子傳輸.

2.1.3 封端、超支化、交聯等策略抑制鏈間相互作用
Chen等[19]提出的聚芴端基聚集機理為聚芴封端以提高光譜穩定性提供了理論依據,封端的分子包括空穴傳輸基團三苯胺[93]以及缺電子的三唑(TAZ)結構28[94];我們組將咔唑作為側基引入剛柔(rodcoil)嵌段共聚芴29[95],促進了其固態時鏈間能量轉移,在空氣中150℃退火2 h,沒有出現長波發射. Gregoriou等[38,96]利用rod-coil嵌段封端方法也得到了穩定的藍光發射,最近,Kwon等[97]研究表明,將聚[五(乙二醇)甲基甲醚]作為位阻封端基團引入(PFO) 30,可以減少基團堆積形成的激基締合物,顯著提高其熱穩定性.
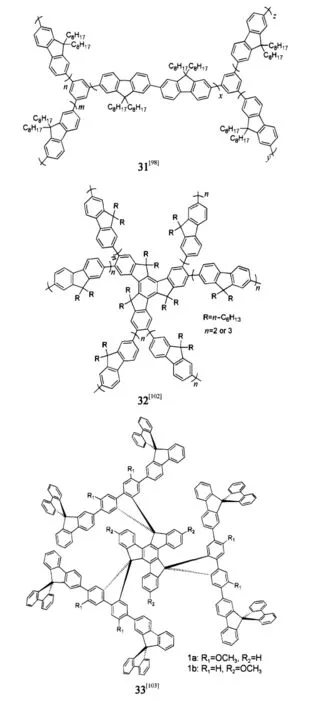
Bo等[98]提出超支化的概念,通過有效的合成手段構建具有獨特分子構型的超支化寡聚芴分子31,可以有效地減弱了聚芴分子間的強的相互作用和分子鏈聚集,引起學術界的關注.我們組[99-102]將超支化應用于提高光譜穩定性,最先設計出具有單分散性的多臂星狀納米尺度聚芴分子32[102],研究表明當支鏈芴的重復單元為2或3時可獲得最穩定的深藍激光器件,發射峰在437 nm,色坐標為(0.15,0.06),固態量子效率達90%.Pei等[103]設計出基于螺芴框架的發散狀三維螺旋立體分子33,其器件發光在7.4 V時亮度為1717 cd·m-2,發射峰在424 nm,色坐標為(0.17,0.08),且具有較高的穩定性.
最早,Miller等[33]在175-200℃下有效引發了苯乙烯基的聚合實現了自交聯34,并指出交聯能夠有效地調控聚芴的超分子堆積結構,從而抑制了鏈間聚集體或激基復合物的形成.我們組[104]設計可光交聯的線性聚芴分子,實現了高量子產率與高光譜穩定性;Ding等[105]合成了主鏈長度為2-4個芴單元、端基含硼酸基團的寡聚芴35,通過在加熱條件下的脫水交聯作用,形成了熱穩定性良好的藍光材料.

隨后,利用多種方式聯合來提高材料穩定性也取得了較好的效果:我們組通過超支化和光交聯結合,得到環氧側鏈修飾的聚芴36[106],有報道采用引入螺環同時交聯的方法來構筑三維立體網絡結構37[107],另外,利用穩定的無機POSS核進行支化結構構筑38[108]也是有效的策略之一.

2.1.4 納米雜化抑制鏈間聚集行為
隨著量子點材料的深入研究和廣泛應用,Wei等[109-110]將CdS、Au量子點引入聚芴,通過設計量子點與樹枝狀聚芴鏈間有效配位作用(圖25),利用量子點三維空間位阻增大鏈間距離,避免聚集,得到高效穩定的薄膜態藍光發射.

圖25 CdS納米晶修飾的樹枝取代聚芴[109]Fig.25 Dendron-substituted polyfluorene modified with CdS nanoparticles[109]
Xu等[111]報道向聚芴體系引入特殊的無機結構基團,如環狀疊氮膦(cyclotriphosphazene)基團,得到相應的有機無機雜化材料39.環狀疊氮膦基團顯著提高聚芴類材料的熱穩定性,并通過立體位阻減少聚集,在200℃熱退火條件下,仍可獲得穩定的藍光發射.
通過巧妙的合成將β-環糊精等大環分子引入聚芴構筑了聚輪烷(polyrotaxanes)(圖26)[112],通過β-環糊精環對聚芴分子線的位阻屏蔽作用,增大鏈間距離,阻隔發色團堆積,也得到了類似的結論.
2.2 避免化學氧化和芴酮缺陷策略
除了上述的從聚集機理出發設計鏈結構抑制聚集等物理缺陷的同時,有些一些文獻針對化學芴酮結構或界面缺陷進行設計,主要包括從材料的合成純化、9-位碳原子替代、位阻胺抗光氧化、構建寬帶隙D-A能量轉移系統以及器件界面優化等方面.

圖26 聚芴鏈與環糊精組成的聚輪烷[112]Fig.26 Polyrotaxanes based on PFO threaded with cyclodextrin[112]
2.2.1 新結構的合成與材料的純化
Holmes等[113]通過合成共聚9-位Si原子取代的芴單體,有效阻止了芴酮結構的生成,得到聚合物40,其發光器件最大發射波長在425 nm,量子效率為62%的穩定深藍光;另外,Holmes等[53]從純化單體,避免單烷基芴缺陷入手,合成無單烷基取代缺陷的聚芴材料,其光致和電致發光光譜均表現出較好的穩定.最近,Lee等[114-115]對芴酮發射的研究表明, LEEB還可能源自芴的9-位碳原子的氧化后,其雜化軌道由sp3轉變為sp2雜化,而導致的芴結構中相連苯環的平面形變.因此以雙鍵將芴的4,5-位固定合成高效穩定的藍光材料PCPP,并合成了相應的材料(BEHP-PCPP)41,得到其熱穩定的發射光譜.
與此同時,我們組[116]采用直接碘化的方法得到了通用的中間體42,然后可以方便的得到含硅、鍺、錫、硼、磷等雜原子的雜芴單體,以此單體為基礎,得到了一系列的雜芴的聚合物;另外,我們組[25,117]還嘗試將傳統抗氧化基——受阻胺五甲基哌啶醇作為側鏈或封端基團引入聚芴體系,如材料43,其熒光發射光譜的穩定性顯著提高,綠光區發射現象明顯減弱.
2.2.2 共聚體系的D-A能量轉移的實現
最近,有人提出設計共聚單元間的能量轉移結構獲得穩定藍光分子.聚芴作為藍光主體發光材料,往往可以通過摻雜,實現處于高能激發態的聚芴發光材料向低能態的客體發光材料的能量轉移,從而阻斷激子被芴酮缺陷捕獲的途徑,以實現理想發光色度與OLED器件的發光效率的提高.能量轉移的實現,要求引入的大基團或阻斷結構同時擔當能量轉移中心,激發態的純聚芴給體分子的發射光譜重疊于功能基團受體分子的吸收光譜,達到有效的F?rster共振能量轉移.Mo等[118]合成了聚3,6-芴的共聚物44,并通過能量轉移策略得到最大發射波長位于420 nm穩定的電致藍光發射光譜.純3,6-聚芴具有3.6 eV的寬帶隙和347 nm的發射波長,其HOMO能級比聚2,7-芴低,為-6.05 eV,表明該材料具有更好的抗氧化能力.
共聚其他類似芴酮的結構如PSO,模擬能量陷落結構是避免LEEB的新策略.Cao等[119]設計共聚2,8-取代或者3,7-取代的穩定高效FSO-PFO藍光材料45,其器件色坐標(0.16,0.07),外量子效率3.6%. PSO有平衡載流子傳輸的作用,并調制能帶2.8至2.9 eV,為深藍光發射.另外,最近Kwon等[120]將烷基芴引入PF的9-位制備材料46,即使發生側鏈氧化生成芴酮結構,也不會影響主鏈發射,減少LEEB.

2.2.3 器件結構優化

為避免陽極中的Ca離子誘導聚芴氧化為酮式結構而出現明顯的綠光發射,Moses等[16]在陽極和發光層之間加入抗氧化緩沖層——電子傳輸材料CF3OXD,可以使材料的熱穩定性和光穩定性都有較大改善,綠光發射現象明顯得以抑制.另外,Neher等[4,121]系統研究和論證了向聚芴發光層摻雜小分子空穴傳輸材料或向器件中加入空穴傳輸層,都可以提高電致發光效率和藍光穩定性.因此,通過摻雜旋涂制備PLED器件是提高聚芴器件性能,避免長波發射的另一種途徑.Jen等[122]制備摻雜寬帶半導體(4.2 eV)的(PVK、PFPE)47材料比例95∶5的旋涂薄膜器件,利用PVK到PFPE的能量轉移,得到外量子效率1.81%,6 V電壓下發射波長397 nm,半峰寬40 nm的穩定藍光材料.

3 聚芴藍光材料穩定性的總結及展望
經過近十年的努力,聚芴材料從器件性能到穩定性方面都得到了大幅度的提高.目前,英國劍橋顯示公司(簡稱CDT)的藍光材料獲得了色坐標(0.15, 0.09)和外量子效率10.4%的摻雜器件.在本文中,我們闡明了LEEB現象產生的兩個重要因素——聚集態和芴酮缺陷,以及二者之間的關系;隨后,就如何從提高材料形態穩定性和環境穩定性策略展開討論.(1)在形態穩定性方面,研究表明聚芴的鏈間ππ堆積對LEEB有較大貢獻,由于π-π堆積要求的作用距離約0.3 nm[41-42,49,58,66],且需要良好的結構匹配,保持合適的鏈間距即可能避免聚集產生的激基締合物發射,并且保持薄膜態載流子的傳輸效率.該過程經歷了從早期研究的基于P-N嵌段的電子結構調制,到通過位阻和構象修飾實現單鏈分子發射.最近與形態穩定性調控相關聯的β相以及微納結構調制成為該領域新的研究熱點.對于摻雜器件的壽命,其相分離形態穩定性問題仍然亟待解決.(2)環境穩定性主要指聚芴對水氧的穩定性,其中,單線態氧可以將芴9-位氧化,因此合成中材料的純化和避免缺陷成為重要課題之一;而氧分子也有猝滅發光的作用.無論是合成無單烷基芴缺陷的單體分子[53],還是在用抗氧化受阻胺消除芴酮[25,117],都沒有完全去除綠光發射,因此環境穩定方面還有很多工作有待材料學家們繼續研究.對于機理研究的模型分子,寡聚物具有高分子的溶液成膜性與小分子的易于純化的特征,成為目前澄清和解決該問題的重要途徑.聚芴類半導體材料的光譜穩定性研究模式為揭示其它共軛聚合物及其退激過程提供了重要參考.
1 Burroughes,J.;Bradley,D.;Brown,A.;Marks,R.;Mackay,K.; Friend,R.;Burns,P.;Holmes,A.Nature,1990,347:539
2 Ohmori,Y.;Uchida,A.;Muro,K.;Yoshino,K.Jpn.J.Appl. Phys.,1991,30:L1941
3 Leclerc,M.J.Polym.Sci.Pol.Chem.,2001,39:2867
4 Neher,D.Macromol.Rapid Commun.,2001,22:1365
5 Lee,J.I.;Klaerner,G.;Miller,R.D.Chem.Mater.,1999,11: 1083
6 Zhu,R.;Feng,J.C.;Huang,W.Chemistry,2005,68:241 [朱 瑞,馮嘉春,黃 維.化學通報,2005,68:241]
7 Tang,C.;Liu,F.;Xu,H.;Huang,W.Prog.Chem.,2007,19:1553 [唐 超,劉 烽,徐 慧,黃 維.化學進展,2007,19:1553]
8 Lai,W.Y.;Mei,Q.B.;Song,J.;Huang,W.J.Nanjing Univ. Posts and Telecom.:Nat.Sci.,2008,28:88 [賴文勇,梅群波,宋 娟,黃 維.南京郵電大學學報:自然科學版,2008,28:88]
9 Chen,Z.Y.;Bai,W.B.;Zhan,C.M.Chin.Polym.Bull.,2007,8: 11 [陳知遠,白衛斌,詹才茂.高分子通報,2007,8:11]
10 Jiang,H.J.;Wan,J.H.;Huang,W.Sci.China,Ser.B:Chem., 2008,51:497 [姜鴻基,萬俊華,黃 維.中國科學B輯:化學,2008,51:497]
11 Zhao,Q.J.;Wu,W.H.Imaging Sci.Photochem.,2008,26:305 [趙前進,吳文輝.影像科學與光化學,2008,26:305]
12 Scherf,U.;Neher,D.Polyfluorene.Berlin:Springer Press,2008: 1-322
13 Pei,Q.B.;Yang,Y.J.Am.Chem.Soc.,1996,118:7416
14 Klaerner,K.;Furhrer,T.;Karg,J.;Chen,W.;Lee,V.;Scott,J.; Miller,R.Macromolecules,1998,31:1099
15 Xie,L.H.;Liu,F.;Tang,C.;Hou,X.Y.;Hua,Y.R.;Fan,Q.L.; Huang,W.Org.Lett.,2006,8:2787
16 Gong,X.O.;Iyer,P.K.;Moses,D.;Bazan,G.C.;Heeger,A.J.; Xiao,S.S.Adv.Funct.Mater.,2003,13:325
17 Sims,M.;Bradley,D.D.C.;Ariu,M.;Koeberg,M.;Asimakis, A.;Grell,M.;Lidzey,D.G.Adv.Funct.Mater.,2004,14:765
18 Weinfurtner,K.H.;Fujikawa,H.;Tokito,S.;Taga,Y.Appl.Phys. Lett.,2000,76:2502
19 Chen,S.H.;Chou,H.L.;Su,A.C.;Chen,S.A.Macromolecules, 2004,37:6833
20 Chen,X.W.;Tseng,H.E.;Liao,J.L.;Chen,S.A.J.Phys.Chem. B,2005,109:17496
21 Montilla,F.;Mallavia,R.Adv.Funct.Mater.,2007,17:71
22 Chen,S.H.;Su,A.C.;Su,C.H.;Chen,S.A.Macromolecules, 2005,38:379
23 Chen,S.H.;Su,A.C.;Chen,S.A.J.Phys.Chem.B,2005,109: 10067
24 Zhao,W.;Cao,T.;White,J.M.Adv.Funct.Mater.,2004,14: 783
25 Hou,X.Y.;Li,T.C.;Liang,J.;Chen,D.Y.;Xie,L.H.;Huang, W.Acta Chim.Sin.,2008,66:2575 [侯曉雅,李廷成,梁 婧,陳道勇,解令海,黃 維.化學學報,2008,66:2575]
26 Klarner,G.;Lee,J.I.;Davey,M.H.;Miller,R.D.Adv.Mater., 1999,11:115
27 Scherf,U.;List,E.J.W.Adv.Mater.,2002,14:477
28 Koizumi,Y.;Seki,S.;Tsukuda,S.;Sakamoto,S.;Tagawa,S. J.Am.Chem.Soc.,2006,128:9036
29 Burnell,T.;Cella,J.A.;Donahue,P.;Duggal,A.;Early,T.; Heller,C.M.;Liu,J.;Shiang,J.;Simon,D.;Slowinska,K.;Sze, M.;Williams,E.Macromolecules,2005,38:10667
30 Bliznyuk,V.;Carter,S.;Scott,J.;Klarner,G.;Miller,R.D. Macromolecules,1999,32:361
31 Bradley,D.D.C.;Grell,M.;Long,X.;Mellor,H.;Grice,A.Proc. SPIE,1998,3145:254
32 Klarner,G.;Davey,M.H.;Chen,W.D.;Scott,J.C.;Miller,R.D. Adv.Mater.,1998,10:993
33 Klarner,G.;Lee,J.I.;Lee,V.Y.;Chan,E.;Chen,J.P.;Nelson, A.;Markiewicz,D.;Siemens,R.;Scott,J.C.;Miller,R.D.Chem. Mater.,1999,11:1800
34 Zeng,G.;Yu,W.L.;Chua,S.J.;Huang,W.Macromolecules, 2002,35:6907
35 Lu,H.H.;Liu,C.Y.;Jen,T.H.;Liao,J.L.;Tseng,H.E.;Huang, C.W.;Hung,M.C.;Chen,S.A.Macromolecules,2005,38: 10829
36 Liao,J.L.;Chen,X.;Liu,C.Y.;Chen,S.A.;Su,C.H.;Su,A.C. J.Phys.Chem.B,2007,111:10379
37 Pei,J.;Liu,X.;Chen,Z.;Zhang,X.;Lai,Y.;Huang,W. Macromolecules,2003,36:323
38 Chochos,C.L.;Kallitsis,J.K.;Keivanidis,P.E.;Baluschev,S.; Gregoriou,V.G.J.Phys.Chem.B,2006,110:4657
39 Qiang,L.L.;Ma,Z.;Zheng,Z.;Yin,R.;Huang,W.Macromol. Rapid Commun.,2006,27:1779
40 Chochos,C.L.;Kallitsis,J.K.;Gregoriou,V.G.J.Phys.Chem. B,2005,109:8755
41 Surin,M.;Hennebicq,E.;Ego,C.;Marsitzky,D.;Grimsdale,A. C.;Mullen,K.;Bredas,J.L.;Lazzaroni,R.;Leclere,P.Chem. Mater.,2004,16:994
42 Surin,M.;Sonar,P.;Grimsdale,A.C.;Mullen,K.;Lazzaroni,R.; Leclere,P.Adv.Funct.Mater.,2005,15:1426
43 List,E.J.W.;Guentner,R.;de Freitas,P.S.;Scherf,U.Adv. Mater.,2002,14:374
44 Gaal,M.;List,E.J.W.;Scherf,U.Macromolecules,2003,36:4236
45 Gamerith,S.;Gaal,M.;Romaner,L.;Nothofer,H.G.;Guntner, R.;de Freitas,P.S.;Scherf,U.;List,E.J.W.Synth.Met.,2003, 139:855
46 Zojer,E.;Pogantsch,A.;Hennebicq,E.;Beljonne,D.;Bredas,J. L.;de Freitas,P.S.;Scherf,U.;List,E.J.W.J.Chem.Phys., 2002,117:6794
47 Franco,I.;Tretiak,S.Chem.Phys.Lett.,2003,372:403
48 Kulkarni,A.P.;Kong,X.X.;Jenekhe,S.A.J.Phys.Chem.B, 2004,108:8689
49 Chi,C.Y.;Im,C.;Enkelmann,V.;Ziegler,A.;Lieser,G.; Wegner,G.Chem.Eur.J.,2005,11:6833
50 Liu,L.L.;Tang,S.;Liu,M.R.;Xie,Z.Q.;Zhang,W.;Lu,P.; Hanif,M.;Ma,Y.G.J.Phys.Chem.B,2006,110:13734
51 Liu,L.L.;Lu,P.;Xie,Z.Q.;Wang,H.P.;Tang,S.;Wang,Z.M.; Zhang,W.;Ma,Y.G.J.Phys.Chem.B,2007,111:10639
52 Liu,L.L.;Yang,B.;Zhang,H.Y.;Tang,S.;Xie,Z.Q.;Wang,H. P.;Wang,Z.M.;Lu,P.;Ma,Y.G.J.Phys.Chem.C,2008,112: 10273
53 Cho,S.Y.;Grimsdale,A.C.;Jones,D.J.;Watkins,S.E.;Holmes, A.B.J.Am.Chem.Soc.,2007,129:11910
54 Grisorio,R.;Suranna,G.P.;Mastrorilli,P.;Nobile,C.F.Adv. Funct.Mater.,2007,17:538
55 Abbel,R.;Wolffs,M.;Bovee,R.;van Dongen,J.;Lou,X.; Henze,O.;Feast,W.;Meijer,E.;Schenning,A.Adv.Mater., 2009,21:597
56 Wang,Z.X.;Shao,H.X.;Ye,J.C.;Zhang,L.;Lu,P.Adv.Funct. Mater.,2007,17:253
57 Tang,S.;Liu,M.R.;Lu,P.;Cheng,G.;Zeng,M.;Xie,Z.Q.;Xu, H.;Wang,H.P.;Yang,B.;Ma,Y.G.;Yan,D.H.Org.Electron., 2008,9:241
58 Liu,L.L.;Qiu,S.;Wang,B.L.;Wang,H.;Xie,Z.Q.;Ma,Y.G. J.Phys.Chem.C,2009,113:5799
59 Wu,Y.S.;Li,J.;Ai,X.C.;Fu,L.M.;Zhang,J.P.;Fu,Y.Q.; Zhou,J.J.;Li,L.;Bo,Z.S.J.Phys.Chem.A,2007,111:11473
60 Ferenczi,T.A.M.;Sims,M.;Bradley,D.D.C.J.Phys.-Condes. Matter,2008,20:045220
61 Zhou,X.H.;Zhang,Y.;Xie,Y.Q.;Cao,Y.;Pei,J. Macromolecules,2006,39:3830
62 Becker,K.;Lupton,J.M.;Feldmann,J.;Nehls,B.S.;Galbrecht, F.;Gao,D.Q.;Scherf,U.Adv.Funct.Mater.,2006,16:364
63 Da Como,E.;Scheler,E.;Strohriegl,P.;Lupton,J.M.;Feldmann, J.Appl.Phys.-Mater.Sci.Proc.,2009,95:61
64 Kappaun,S.;Scheiber,H.;Trattnig,R.;Zojer,E.;List,E.J.W.; Slugovc,C.Chem.Commun.,2008:5170
65 Sainova,D.;Miteva,T.;Nothofer,H.;Scherf,U.;Glowacki,I.; Ulanski,J.;Fujikawa,H.;Neher,D.Appl.Phys.Lett.,2000,76: 1810
66 Kulkarni,A.P.;Jenekhe,S.A.Macromolecules,2003,36:5285
67 Setayesh,S.;Grimsdale,A.C.;Weil,T.;Enkelmann,V.;Müllen, K.;Meghdadi,F.;List,E.J.W.;Leising,G.J.Am.Chem.Soc., 2001,123:946
68 Chou,C.H.;Shu,C.F.Macromolecules,2002,35:9673
69 Marsitzky,D.;Vestberg,R.;Blainey,P.;Tang,B.T.;Hawker,C. J.;Carter,K.R.J.Am.Chem.Soc.,2001,123:6965
70 Wu,C.W.;Tsai,C.M.;Lin,H.C.Macromolecules,2006,39: 4298
71 Fu,Y.Q.;Li,Y.;Li,J.;Yan,S.;Bo,Z.S.Macromolecules,2004, 37:6395
72 Yu,W.L.;Pei,J.;Huang,W.;Heeger,A.J.Adv.Mater.,2000, 12:828
73 Zhu,R.;Wen,G.A.;Feng,J.C.;Chen,R.F.;Zhao,L.;Yao,H. P.;Fan,Q.L.;Wei,W.;Peng,B.;Huang,W.Macromol.Rapid Commun.,2005,26:1729
74 Wu,F.I.;Dodda,R.;Jakka,K.;Huang,J.H.;Hsu,C.S.;Shu,C. F.Polymer,2004,45:4257
75 Su,H.J.;Wu,F.I.;Shu,C.F.Macromolecules,2004,37:7197
76 Wu,Y.G.;Li,J.;Fu,Y.Q.;Bo,Z.S.Org.Lett.,2004,6:3485
77 Tseng,Y.H.;Shih,P.I.;Chien,C.H.;Dixit,A.K.;Shu,C.F.; Liu,Y.H.;Lee,G.H.Macromolecules,2005,38:10055
78 Tang,S.;Liu,M.;Lu,P.;Xia,H.;Li,M.;Xie,Z.Q.;Shen,T.Z.; Gu,C.;Wang,H.P.;Yang,B.;Ma,Y.G.Adv.Funct.Mater., 2007,17:2869
79 Wu,Y.G.;Zhang,J.Y.;Bo,Z.S.Org.Lett.,2007,9:4435
80 Horhant,D.;Liang,J.J.;Virboul,M.;Poriel,C.;Alcaraz,G.; Rault-Berthelot,J.Org.Lett.,2006,8:257
81 Poriel,C.;Liang,J.J.;Rault-Berthelot,J.;Barriere,F.;Cocherel, N.;Slawin,A.M.Z.;Horhant,D.;Virboul,M.;Alcaraz,G.; Audebrand,N.;Vignau,L.;Huby,N.;Wantz,G.;Hirsch,L. Chem.Eur.J.,2007,13:10055
82 Poriel,C.;Rault-Berthelot,J.;Barriere,F.;Slawins,A.Org.Lett., 2008,10:373
83 Cocherel,N.;Poriel,C.;Rault-Berthelot,J.;Barriere,F.; Audebrand,N.;Slawin,A.M.Z.;Vignau,L.Chem.Eur.J.,2008, 14:11328
84 Yu,W.L.;Pei,J.;Cao,Y.;Huang,W.;Heeger,A.J.Chem. Commun.,1999:1837
85 Yu,W.L.;Cao,Y.;Pei,J.;Huang,W.;Heeger,A.J.Appl.Phys. Lett.,1999,75:3270
86 Wu,F.I.;Dodda,R.;Reddy,D.S.;Shu,C.F.J.Mater.Chem., 2002,12:2893
87 Wong,W.Y.;Liu,L.;Cui,D.M.;Leung,L.M.;Kwong,C.F.; Lee,T.H.;Ng,H.F.Macromolecules,2005,38:4970
88 Kulkarni,A.P.;Zhu,Y.;Jenekhe,S.A.Macromolecules,2005, 38:1553
89 Grigalevicius,S.;Ma,L.;Xie,Z.Y.;Scherf,U.J.Polym.Sci.Pol. Chem.,2006,44:5987
90 Wang,H.Y.;Feng,J.C.;Wen,G.A.;Jiang,H.J.;Wan,J.H.; Zhu,R.;Wang,C.M.;Wei,W.;Huang,W.New J.Chem.,2006, 30:667
91 Liu,X.M.;Xu,J.W.;Lu,X.H.;He,C.B.Macromolecules, 2006,39:1397
92 Zhou,X.H.;Niu,Y.H.;Huang,F.;Liu,M.S.;Jen,A.K.Y. Macromolecules,2007,40:3015
93 Miteva,T.;Meisel,A.;Knoll,W.;Nothofer,H.G.;Scherf,U.; Muller,D.C.;Meerholz,K.;Yasuda,A.;Neher,D.Adv.Mater., 2001,13:565
94 Hung,M.C.;Liao,J.L.;Chen,S.A.;Chen,S.H.;Su,A.C. J.Am.Chem.Soc.,2005,127:14576
95 Lu,S.;Liu,T.X.;Ke,L.;Ma,D.G.;Chua,S.J.;Huang,W. Macromolecules,2005,38:8494
96 Chochos,C.L.;Tsolakis,P.K.;Gregoriou,V.G.;Kallitsis,J.K. Macromolecules,2004,37:2502
97 Kwon,Y.K.;Kim,H.S.;Kim,H.J.;Oh,J.H.;Park,H.S.;Ko,Y. S.;Kim,K.B.;Kim,M.S.Macromolecules,2009,42:887
98 Li,J.;Bo,Z.S.Macromolecules,2004,37:2013
99 Xin,Y.;Wen,G.A.;Zeng,W.J.;Zhao,L.;Zhu,X.R.;Fan,Q. L.;Feng,J.C.;Wang,L.H.;Wei,W.;Peng,B.;Cao,Y.;Huang, W.Macromolecules,2005,38:6755
100 Lai,W.Y.;He,Q.Y.;Zhu,R.;Chen,Q.Q.;Huang,W.Adv. Funct.Mater.,2008,18:265
101 Wen,G.;Xin,Y.;Zhu,X.;Zeng,W.;Zhu,R.;Feng,J.;Cao,Y.; Zhao,L.;Wang,L.;Wei,W.;Peng,B.;Huang,W.Polymer, 2007,48:1824
102 Lai,W.Y.;Xia,R.D.;He,Q.Y.;Levermore,P.A.;Huang,W.; Bradley,D.D.C.Adv.Mater.,2009,21:355
103 Luo,J.;Zhou,Y.;Niu,Z.;Zhou,Q.;Ma,Y.;Pei,J.J.Am.Chem. Soc.,2007,129:11314
104 Yuan,X.D.;Wen,G.A.;Qi,X.Y.;Tang,D.F.;Peng,B.;Wang, L.H.;Wei,W.;Huang,W.Acta Polym.Sin.,2006:1029 [袁翔東,溫貴安,戚筱英,湯多峰,彭 波,汪聯輝,韋 瑋,黃 維.高分子學報,2006:1029]
105 Li,Y.N.;Ding,J.F.;Day,M.;Tao,Y.;Lu,J.P.;D′Iorio,M. Chem.Mater.,2003,15:4936
106 Tang,D.;Wen,G.;Qi,X.;Wang,H.;Peng,B.;Wei,W.;Huang, W.Polymer,2007,48:4412
107 Marsitzky,D.;Murray,J.;Scott,C.;Carter,K.R.Chem.Mater., 2001,13:4285
108 Lin,W.;Chen,W.;Wu,W.;Niu,Y.;Alex,K.Macromolecules, 2004,37:2335
109 Chou,C.;Wang,H.;Wei,K.;Huang,J.Adv.Funct.Mater., 2006,16:909
110 Hsu,S.L.;Chou,C.H.;Chen,C.P.;Wei,K.H.Adv.Funct. Mater.,2007,17:2534
111 Xu,J.W.;Toh,C.L.;Ke,K.L.;Li,J.J.;Cho,C.M.;Lu,X.H.; Tan,E.W.;He,C.B.Macromolecules,2008,41:9624
112 Cacialli,F.;Wilson,J.;Michels,J.;Daniel,C.;Silva,C.;Friend, R.;Severin,N.;Samorì,P.;Rabe,J.;O′Connell,M.Nat.Mater., 2002,1:160
113 Chan,K.L.;McKiernan,M.J.;Towns,C.R.;Holmes,A.B. J.Am.Chem.Soc.,2005,127:7662
114 Park,S.H.;Jin,Y.;Kim,J.Y.;Kim,S.H.;Kim,J.;Suh,H.;Lee, K.Adv.Funct.Mater.,2007,17:3063
115 Kim,J.;Jin,Y.;Song,S.;Kim,S.H.;Park,S.H.;Lee,K.;Suh,H. Macromolecules,2008,41:8324
116 Chen,R.;Fan,Q.;Zheng,C.;Huang,W.Org.Lett.,2006,8:203 117 Si,S.M.;Xie,L.H.;Wei,W.;Peng,B.;Huang,W.Acta Polym. Sin.,2007:148 [司三民,解令海,韋 瑋,彭 波,黃 維.高分子學報,2007:148]
118 Mo,Y.Q.;Jiang,X.;Cao,D.R.Org.Lett.,2007,9:4371
119 Li,Y.Y.;Wu,H.B.;Zou,J.H.;Ying,L.;Yang,W.;Cao,Y.Org. Electron.,2009,10:901
120 Jin,J.K.;Kwon,S.K.;Kim,Y.H.;Shin,D.C.;You,H.;Jung,H. T.Macromolecules,2009,42:6339
121 Yang,X.H.;Jaiser,F.;Neher,D.;Lawson,P.V.;Brdéas,J.L.; Zojer,E.;Güntner,R.;deFreitas,P.S.;Forster,M.;Scherf,U. Adv.Func.Mater.,2004,14:1097
122 Huang,F.;Niu,Y.H.;Liu,M.S.;Zhou,X.H.;Tian,Y.Q.;Jen, A.K.Y.Appl.Phys.Lett.,2006,89:081104
June 1,2009;Revised:November 13,2009;Published on Web:February 23,2010.
Spectral Stability of Polyfluorene-Based Semiconductors
LIANG Jing1,2QIAN Yan1,2XIE Ling-Hai1,2,*SHI Nai-En1,2CHEN Shu-Fen1,2DENG Xian-Yu1,2HUANG Wei1,2,*
(1Key Laboratory for Organic Electronics&Information Displays,Nanjing 210046;2Institute of Advanced Materials,Nanjing University of Posts and Telecommunication,Nanjing 210046,P.R.China)
The physical and chemical properties of organic semiconductors play key roles in the performance of optoelectronic devices.The manipulation of these properties offers research opportunities and challenges in physical chemistry.The spectral stability and the origin of the low-energy emission band(LEEB)in polyfluorenes-based blue light-emitting diodes have
much attention over the past few decades.In this review,we categorized various LEEB phenomena according to characterization and related mechanisms,including inter-chain aggregates and/or excimers,on-chain ketone defect emissions,interchain ketone-based excimers,and hydroxy-terminated oxidation on the interface of devices.Recent advances in highly stable blue light-emitting polyfluorenes are categorically summarized.This review highlighted the contributions of the steric hindrance effects of various nonplanar bulky groups,molecular conformations and topologies of chains and antioxidant hindered amine light stabilizers(HALS), together with physical blending as well as interface engineering.
Electroluminescent;Conjugated polymer;Polyfluorene;Low-energy emission band;Spectrum stability; Blue light-emitting semiconductor
O649
*Corresponding authors.Email:iamlhxie@njupt.edu.cn,iamwhuang@njupt.edu.cn;Tel:+86-25-85866008.
The project was supported by the National Key Basic Research Program of China(973)(2009CB930600),National Natural Science Foundation of China(20774043,20704023,60876010,60706017,60907047),Key Project of Chinese Ministry of Education(707032,104246,208050),Natural Science Foundation of Jiangsu Province,China(BK2008053,BK2009423,08KJD150016,08KJB510013,09KJB150009,SJ209003,TJ207035),
and NJUPT(NY207042,NY207041,NY207038).
國家重點基礎研究發展規劃(973)項目(2009CB930600),國家自然科學基金(20774043,20704023,60876010,60706017,60907047),教育部重大培育基金項目(707032),教育部重點項目(104246,208050),江蘇省基礎研究計劃(自然科學基金)(BK2008053,BK2009423),江蘇省高校自然科學基礎研究(08KJD150016,08KJB510013,09KJB150009,SJ209003),江蘇省高等學校優秀科技創新團隊(TJ207035),南京郵電大學引進人才啟動基金(NY207042,NY207041,NY207038)資助
黃維,于1979年進入北京大學化學系學習并先后獲得理學學士、碩士和博士學位.
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
哲學評論(2021年2期)2021-08-22 01:53:34
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
汽車工程學報(2017年2期)2017-07-05 08:13:02
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
