鐵質原料主要化學成分的測定淺析
2010-12-26 06:32:10李筠樂羅曄
四川水泥 2010年2期
關鍵詞:影響
李筠樂 羅曄
(河南省水泥質量監督檢驗站,鄭州市450053)

0 前 言
鐵質校正原料是用以補充配合生料中氧化鐵不足的原料,要求鐵質原料中氧化鐵含量一般不低于40%。通常采用的是硫酸工業廢渣——硫鐵礦渣(俗稱鐵粉),即硫鐵礦經過煅燒脫硫以后排出的粉狀殘渣。其主要礦物成分有赤鐵礦、針鐵礦、纖鐵礦等,氧化鐵的含量大于50%。許多水泥企業目前使用較多的鐵質原料是硫酸渣,其次是鐵礦石、銅礦渣或鉛礦渣等。合理地使用鐵質原料能提高熟料強度,降低熟料燒成熱耗并提高窯的產量。
我們對水泥生料進行配料計算前,必須要做鐵質原料的化學全分析。本文將硅鐵鋁的分析列為鐵質原料全分析的重點。因為鐵的含量很高,通常采用鉍鹽返滴定法。鋁的測定,為防止高含量鐵以及鈦元素的干擾,采用氟化銨置換法。硅采用改進的氟硅酸鉀容量法進行檢測。我們通過分析實驗對鐵質原料的全分析總結了相關的技術要點。
1 試樣的分解及試驗溶液的制備
目前用氫氧化鈉作熔劑以銀坩堝為熔器進行鐵質原料分析是行之有效的,此方法測定程序有很大簡化,并且準確度較高。以鐵礦石為例,用氫氧化鈉為熔劑的熔融條件見表1。

表1 氫氧化鈉作熔劑時試樣的熔融條件
上述熔融條件下制備的溶液冷卻后移入250ml容量瓶,用水稀釋至標線搖勻。該溶液供測定SiO2、Fe2O3、Al2O3、CaO、MgO用。
2 SiO2的測定
我們在用氫氧化鈉為熔劑,銀坩鍋為熔器的熔融條件下制備的溶液測定SiO2時,通過大量的實驗對比研究,采用改進的氟硅酸鉀容量法快速測定硅。該方法與傳統的過飽和沉淀—小體積中和—氟硅酸鉀容量法相比較,可概括為近飽和沉淀—大體積中和—氟硅酸鉀容量法。
2.1 SiO2代用法在具體操作上的改進
(1)氯化鉀無需加到溶液飽和并過量,而是與氟化鉀同時定量加入到經堿熔并剛剛用硝酸溶解的熱試樣溶液中。
(2)對溶液進行水冷的同時,通過對溶液的攪拌,使氟硅酸鉀沉淀快速完全地生成。
(3)由于無氯化鉀晶體析出,對氟硅酸鉀沉淀的過濾與洗滌快速而簡單。有效地減少了沉淀的溶解與水解。
(4)采用較大的氯化鉀—乙醇中和液體積,提高了中和殘余酸終點時中和液中乙醇的有效濃度,從而減小了沉淀的溶解與水解。
(5)采用甲基紅為中和殘余酸滴定終點的指示劑,使終點與化學計量點更相近。同時,終點pH小于化學計量點的pH值,有利于減小氟硅酸鉀對滴定終點的影響,終點變色敏銳。
(6)在滴定氫氟酸的過程中,由于甲基紅的存在,可以預指示終點的到達,使分析者觀察終點時精力集中,有利于減少滴定誤差。
2.2 兩個滴定過程選擇不同指示劑的理論分析
中和殘余酸的過程實際上是用氫氧化鈉標準滴定溶液滴定有少量氟共存的硝酸;在中和完殘余酸的沉淀中,加入沸水則使沉淀溶解,并水解產生氫氟酸。因此此時的滴定仍然是強堿對氫氟酸的滴定。但其化學計量點不同于對單純氫氟酸的滴定,因為此時氫離子的濃度不僅受氫氟酸解離常數的限制,而且還要受SiF62-水解常數的影響,總的解離常數為SiF62-水解常數與氫氟酸解離常數之積,即此時的滴定體系等價于強堿對一種較HF弱的酸的滴定,因而其滴定曲線突躍減小,等當點pH增高。因此中和殘余酸和滴定水解生成的HF,雖然都是用氫氧化鈉滴定氫氟酸,但由于滴定體系中共存物質的不同及介質與介質溫度的不同,其滴定曲線及等當點是不同的,指示其終點的最佳指示劑也不盡相同。
中和殘余酸滴定曲線和滴定水解HF滴定曲線分別見圖1和圖2。指示劑變色點pH值見表2。
由圖1和圖2中的滴定曲線比較可以看出:兩條滴定曲線是有顯著差異的。中和殘余酸時,曲線的突躍范圍為pH5~9,化學計量點為7.0,屬強堿滴定強酸型;滴定水解HF時,曲線突躍范圍很不明顯,化學計量點約為7.5,屬強堿滴定弱酸型。

圖1 中和殘余酸滴定曲線

圖2 滴定水解HF滴定曲線

表2 指示劑變色點pH值
根據表2中各指示劑變色點數據,應選擇甲基紅作滴定殘余酸的指示劑;選擇酚酞作滴定水解HF的指示劑。
2.3 洗滌與中和過程中氟硅酸鉀的溶解
在一定的酸度下,增加鉀離子的濃度和適量增加氟離子的濃度有利于氟硅酸鉀沉淀生成完全;在對沉淀的洗滌過程中,由于氟離子濃度的降低,沉淀的水解和溶解是不可避免的,水解平衡時,沉淀的水解量可達到21%,因而,縮短洗滌時間,使水解反應來不及進行,是減小沉淀水解的有效方法。在中和殘余酸的過程中,由于氫離子濃度的減小,使SiF62-水解的趨勢更大。在中和液中加入乙醇可以從三個方面抑制沉淀的溶解:
(1)降低氟硅酸鉀沉淀的溶解度,實驗表明:50%乙醇溶液能使氟硅酸鉀沉淀的溶解度減小1000倍;
(2)減小水解常數,有效水濃度的降低,能使SiF62-的水解常數減小;
(3)降低水解速度,實驗表明,氟硅酸鉀在50%乙醇溶液中也能逐漸水解,但速度要比在水中慢得多。
3 Fe2O3的測定
以磺基水楊酸鈉為指示劑,用EDTA直接滴定鐵,由于指示劑靈敏度低,在終點到達之前紅色就已消退,其回收率總是低于100%,相對誤差可達到1%以上,對于生料水泥等鐵含量低的樣品影響不大,但對于鐵礦石等高鐵含量的樣品,影響就十分顯著了。同時由于鐵與EDTA生成的配合物呈深黃色,使滴定終點很難判斷,因而用EDTA直接滴定法測Fe2O3的結果誤差較大。用過量的EDTA與鐵完全配合,然后以半二甲酚橙為指示劑,鉍鹽回滴過量的EDTA,終點變色敏銳,對高含量和低含量的鐵均可進行完全回收,見表3。
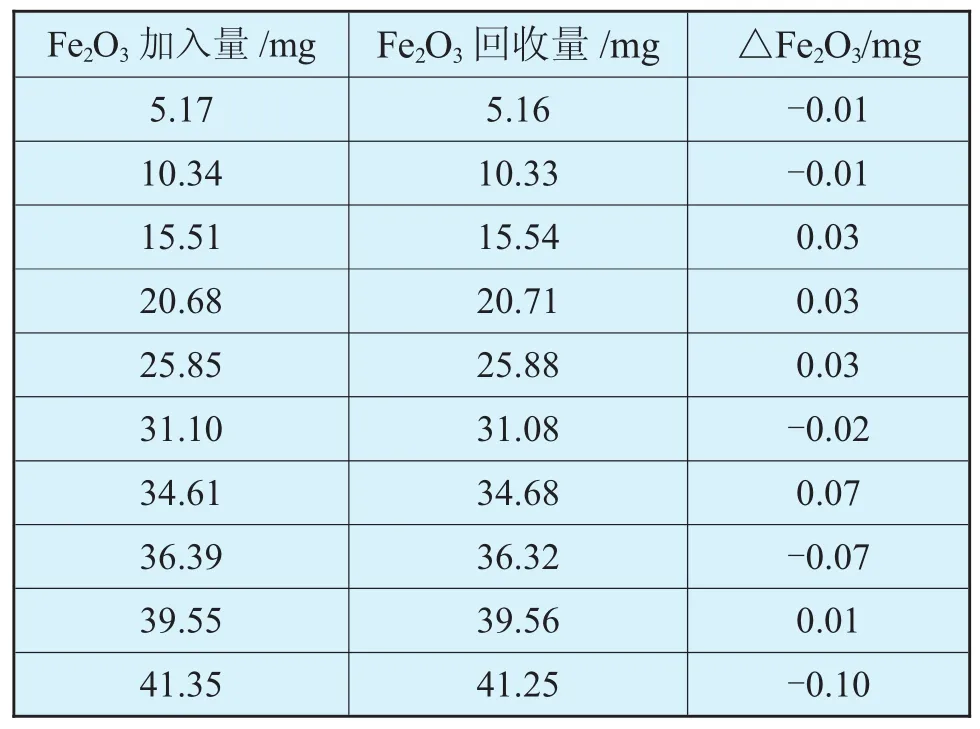
表3 EDTA-鉍鹽回滴法對純鐵標準溶液的回收結果
影響鉍鹽返滴定的因素我們進行了如下分析。
3.1 氯離子對滴定的影響
(1)由于在鹽酸介質中,Cl-與滴定劑Bi3+可生成BiOCl沉淀,從而影響終點的觀察,大量Cl-的存在常會引起較大的負偏差。
(2)實驗證明,當實驗溶液體積為200ml時,允許有相當于1ml濃鹽酸的Cl-存在,對測定結果無影響。但終點變化稍受干擾,因此在用銀坩堝熔樣時,應盡量減少洗滌銀坩堝的稀鹽酸用量。
HCl對滴定的影響見表4。
鋁離子共存時溶液pH值和EDTA標準滴定溶液的過量量對測定結果的影響見表5。
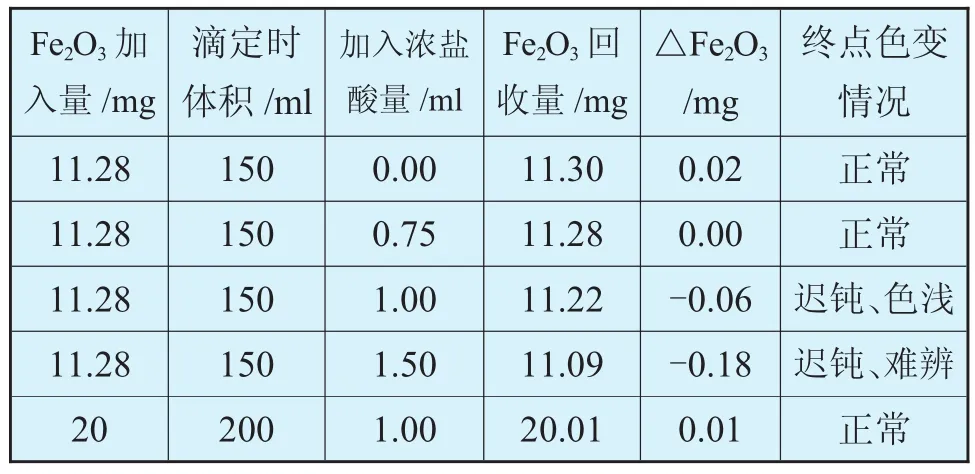
表4 HCl對滴定的影響
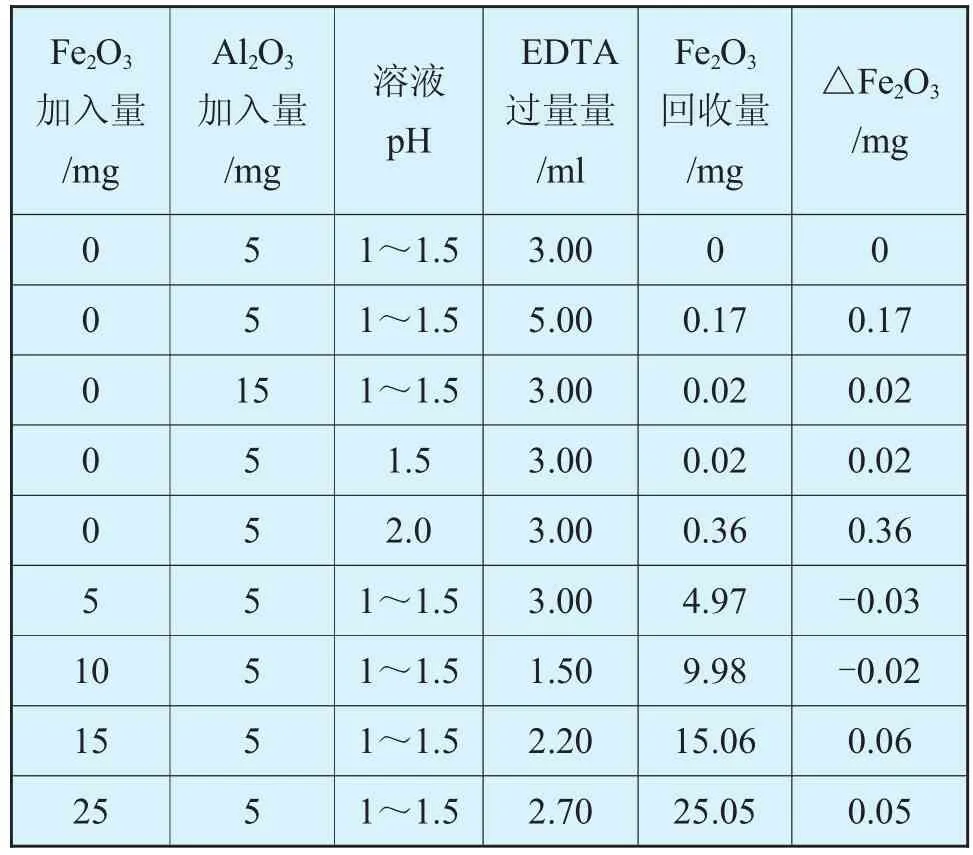
表5 鋁離子共存時溶液pH值和EDTA標準滴定溶液的過量量對測定結果的影響
實驗結果表明:在鋁離子存在時,滴定鐵時pH越低,鋁對其影響越小,但pH<1.0時,終點變化已不明顯,反應遲鈍,當pH在1.0~1.5的范圍內,鋁離子基本不干擾測定,終點明顯;當0.015mol/L EDTA超過5mL時,滴定終點易返色、拖長,測定結果偏高。一般將0.015mol/LEDTA控制在3mL以內比較適當。控制的方法是首先向滴定溶液中加入1~2滴磺基水楊酸鈉指示劑,然后用0.015mol/L EDTA緩慢滴定至紅色消退再過量1~2ml即可。
另外,當有鋁離子存在時,加入EDTA后放置的時間及溶液溫度對測定結果也有一定的影響。放置時間太短,溶液溫度太低,則鐵離子難以與EDTA完全配位使測定結果偏低;反之,放置時間太長,溶液溫度太高,則少量鋁離子可能與EDTA配位使測定結果偏高。試驗表明,放置1~3分鐘,溶液溫度在20~35℃時,測定的結果穩定,效果較好。
3.2 其它因素對滴定的影響
(1)進行鐵鋁鈦連續測定時,滴鐵后應立即向溶液中加入0.2~0.5mLEDTA,使溶液變為黃色,否則容易封閉指示劑。
(2)用鉍鹽標準滴定溶液返滴定時,滴定速度應緩慢,且不斷攪拌,終點為淺橙紅色且穩定即可。不要將終點顏色滴定至深橙紅色或紅色,否則測定結果嚴重偏低。
(3)半二甲酚橙指示劑質量不好時終點不敏銳,應購買質量好的半二甲酚橙指示劑。配置好的半二甲酚橙指示劑溶液不宜久放,否則會導致靈敏度下降。
4 Al2O3的測定
硫酸銅回滴法測定生料、熟料及水泥中的Al2O3已得到廣泛應用,但用此法測定高鐵試樣中的Al2O3,則需通過二次沉淀才能將鐵與鋁鈦分離。由于過程繁瑣,我們采用氟化銨置換法測鐵質原料中的Al2O3。
氟化銨置換法由于稱樣量少(30毫克),相對滴定誤差較大。每滴滴定劑的誤差大于0.12%。本方法對化驗員的經驗要求較高,操作的技術難度較大。屬傳統的方法,定為仲裁法。該法的操作要點:
(1)氟離子能與鋁、鉛和鈦等生成比EDTA更穩定的配合物。
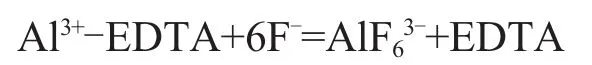
(2)預先加入過量的EDTA與鐵、鋁、鈦、錳等離子配位生成配合物。
(3)加入苦杏仁酸與鈦生成配合物,使鈦無法進一步與后加入的F-反應,以保證游離出的EDTA僅是與原鋁離子配位。10mL100g/L苦杏仁酸溶液可消除試樣中2%~5%的TiO2的干擾。
(4)以半二甲酚橙為指示劑,以鉛鹽溶液滴定剩余的EDTA恰至終點,此時溶液中已無游離的EDTA存在,鋁離子與錳離子等均與EDTA定量配位。因尚未加入氟化銨進行置換,故此時不必記錄鉛鹽溶液的消耗數。
(5)第一次用鉛鹽溶液滴定至終點后,要立即加入氟化銨溶液且加熱進行置換,否則痕量的鈦會與半二甲酚橙指示劑配位,形成穩定的橙紅色配合物,影響第二次滴定。
(6)氟化銨的加入量不宜過多,因為大量的氟化物可與鐵離子反應,造成誤差。一般分析中,100mg以內的三氧化二鋁加1g氟化銨(或10ml100g/l的溶液)可完全滿足置換反應的需要。
(7)用氟化銨置換法測得的氧化鋁是純氧化鋁含量,不受測定鐵鈦滴定誤差的影響,結果穩定,
5 CaO和MgO的測試
采用EDTA配位滴定法測定鈣、鎂時,由于鐵的含量高,掩蔽劑的掩蔽效果受到限制,使測定終點變色遲鈍,不易觀察,甚至無法觀察。因此,將試液中的鐵沉淀并分離除去,問題就迎刃而解了。
采用氨水沉淀分離鐵鋁鈦是經典方法,操作方法十分成熟。試驗表明:氨水不僅可將鐵鋁鈦等金屬離子沉淀完全,而且溶液中的硅酸也能基本沉淀,滴定鈣、鎂時,無需加入氟化鉀對硅進行掩蔽。
由于沉淀鐵鋁鈦時加入甲基紅指示劑會影響滴定鈣、鎂時終點的觀察,因此當鐵沉淀完全時,可能已有部分鋁生成AlO2-而溶解,為了消除其影響,在滴定鈣鎂時,應加入5ml三乙醇胺掩蔽。一般鐵礦石中鈣鎂含量都較低,用熱水沖洗沉淀3~5次即可將沉淀中的鈣鎂離子洗凈。
6 燒失量
相對于硅鐵鋁,燒失量的化學分析較為簡單。我們分別從灼燒溫度、高溫爐的發熱介質、盛樣器皿、灼燒時間、升溫制度等試驗條件上作出總結:
實驗結果表明:溫度對燒失量的影響最明顯,950℃下的燒失量明顯高于800℃下的燒失量;高溫爐的發熱介質對燒失量結果也有較大的影響,這是因為發熱介質為電爐絲,爐膛內氣氛為氧化氣氛,可使低價元素氧化而使樣品增重,而硅碳棒爐為還原性氣氛,硫化物等組份難以氧化而揮發使燒失量增加;灼燒時間對燒失量較高的樣品有一定的影響;升溫制度對結果的影響較小。
由于鐵礦石在高溫下灼燒時,存在著較明顯的氧化和分解化學反應,故其燒失量與灼燒條件有著密切的關系,因而固定燒失量測定的條件是十分必要的。實驗表明:用瓷坩堝盛放樣品,放入已恒溫到950℃的電阻絲高溫爐中,易得到較穩定的結果。
7 結 論
(1)測定各成份時所用的儀器、器皿具有較高的精密度,在分析速度和易操作性方面,有了較大的提高,并保證了分析結果的準確性和重現性。
(2)采用“近飽和沉淀-大體積中和-氟硅酸鉀容量法”測定SiO2,較常規的容量快速分析方法精度提高2倍;操作的技術難度也大為降低。
(3)測定CaO和MgO時,先用氨水沉淀硅、鐵、鋁、鈦等干擾元素,使滴定終點變色敏銳,減小終點誤差。
(4)試樣中常含有多種有色金屬元素,可能對待測金屬元素的測定產生干擾。
猜你喜歡
中學生數理化·八年級物理人教版(2022年3期)2022-03-16 05:55:08
當代陜西(2021年2期)2021-03-29 07:41:24
家庭影院技術(2020年10期)2020-12-14 07:54:18
媽媽寶寶(2017年3期)2017-02-21 01:22:28
中國塑料(2016年3期)2016-06-15 20:30:00
通信電源技術(2016年3期)2016-03-26 07:13:38
知識經濟·中國直銷(2016年3期)2016-02-27 16:15:49
現代檢驗醫學雜志(2014年6期)2014-02-02 03:02:04
閱讀與作文(小學低年級版)(2011年3期)2011-01-01 00:00:00
