嗎啡依賴大鼠不同腦區肌動蛋白及其結合蛋白的表達變化
2011-05-31 08:49:10閆彩珍郝曉玲王天魁
中國藥理學通報 2011年8期
關鍵詞:海馬
閆彩珍,于 亮,劉 川,郝曉玲,王天魁
(河北醫科大學藥理學教研室,河北石家莊 050017)
阿片類藥物成癮的主要特征表現為強迫性和持續性藥物使用。目前研究認為藥物成癮行為的持久存在,可能與轉錄因子短暫調節所致的突觸結構及相關神經回路的結構重塑有關[1]。成癮藥物所致持續性結構改變的一個備受關注的例子是,反復使用成癮藥物可引起與情感動機、獎賞和學習有關腦區神經元樹突棘的結構產生持久改變[2],提示突觸聯系發生了可塑性變化。
樹突棘是神經元之間形成興奮性突觸的位置,它們在神經元中呈現不同的形狀,目前已證實樹突棘形態動力學是由肌動蛋白動態調節的,肌動蛋白在棘突形態形成中發揮重要作用[3]。肌動蛋白是真核細胞最主要的骨架成份之一,它以單體(G-actin)和纖維狀(F-actin)兩種形式存在,并且單體與纖維狀肌動蛋白之間可以相互轉換。在神經元樹突棘可以觀察到與肌動蛋白有關的運動性,熒光漂白恢復技術分析顯示樹突棘F-actin可以發生快速的循環。藥理學研究顯示,控制肌動蛋白循環可以導致樹突棘發生形態學改變,提示肌動蛋白在調節神經元樹突棘的形態和動力學中發揮重要作用[4]。研究顯示,長期嗎啡處理可增加伏隔核F-actin的含量[5],條件性位置厭惡大鼠杏仁核和背側海馬肌動蛋白發生重構[6],但是對嗎啡依賴不同時程大鼠不同腦區肌動蛋白的重構并不清楚,同時嗎啡影響細胞骨架肌動蛋白表達及重構的分子機制也尚未闡明。
本研究擬從影響神經元形態和功能的骨架肌動蛋白入手,采用動物行為學、Western blot等方法,研究嗎啡依賴大鼠部分腦區肌動蛋白及其結合蛋白p-cofilin的表達變化,探討骨架肌動蛋白結合蛋白在嗎啡誘導的神經元突觸可塑中的作用,進一步深化阿片類藥物成癮的分子生物學機制。
1 材料與方法
1.1 材料
1.1.1動物♂ Sprauge Dawley(SD)大鼠,體質量(200±20)g,購自河北省實驗動物中心。
1.1.2藥物及試劑鹽酸嗎啡注射液購自沈陽第一制藥廠;鹽酸納洛酮注射液購自北京華素制藥股份有限公司;Actin抗體購自美國Santa Cruz公司;pcofilin抗體購自美國Cell Signaling公司;BCA蛋白定量試劑盒購自美國PIERCE公司;辣根過氧化酶標記的山羊抗小鼠、羊抗兔抗體購自美國KPL公司;DAB顯色試劑盒購自北京中山生物技術有限公司。
1.1.3主要儀器5417R型低溫離心機,德國Eppendorf公司;超高速離心機,美國 Beckman公司;AG135型電子天平,瑞士Mettler Toledo公司;1004型恒溫水浴箱,德國GFL公司;SHY-200B型水平搖床,德國Leica公司;Millipore純水儀,美國Millipore公司;DYY-11型多用電泳儀、DYC-Z22A型電泳槽,北京六一儀器廠;凝膠成像儀,美國Themo Foma公司。
1.2 方法
1.2.1大鼠嗎啡依賴模型建立♂ SD大鼠42只,隨機分為嗎啡依賴1周組、嗎啡依賴2周組和嗎啡依賴4周組,各依賴組均設對照。每組7只大鼠,分籠喂養,自由進食、飲水,動物適應新環境3 d后進行實驗。采用腹腔遞增注射鹽酸嗎啡建立嗎啡依賴模型,連續注射7 d,每天3次,劑量按5,10,15,20,30,40,50 mg·kg-1逐日遞增,對照組給予同體積生理鹽水。d 8,在各組大鼠中每組隨機選取2只,于嗎啡注射前2 h皮下注射鹽酸納洛酮5 mg·kg-1,觀察戒斷癥狀,并進行評分,判斷大鼠嗎啡依賴模型是否建立成功。然后將嗎啡依賴1周組與對照組大鼠斷頭處死,分離相關腦區。嗎啡依賴2周組、4周組繼續分別給予50 mg·kg-1鹽酸嗎啡,每天1次,連續注射1或3周。在最后一次給藥后5 h斷頭處死大鼠,迅速分離前額葉皮層、丘腦、海馬及紋狀體,置液氮快速冷凍后轉移至-80℃冰箱保存。
1.2.2戒斷癥狀評分標準[7]① 跳躍、濕狗樣抖動、扭體、搖頭、打哈欠、掃尾:0分=無;1分=1~5次;2分=6~10次;3分 >10次。② 齒顫、咀嚼(次與次之間至少間隔30 s):0分=無;1分=1~10次;2分=11~20次;3分>20次。③ 流涎、流淚、豎毛、眼瞼下垂、激惹和腹瀉:0分=無;1分=輕度;2分=中度;3分=明顯。
1.2.3腦組織總蛋白的制備腦組織按每100 mg組織加入1 ml的組織蛋白裂解液(50 mmol·L-1Tris-Cl,pH 7.4,150 mmol·L-1NaCl,1 mmol·L-1EGTA,1 mmol·L-1EDTA,1%NP40,1 mmol·L-1DTT,1 mmol·L-1PMSF,1 mg·L-1aprotintin,1 mg·L-1pepstatin A,1 mg·L-1leupeptin),在 DY-89Ⅰ型電動勻漿器下勻漿30 s,4℃ 12 000 r·min-1離心20 min,收集上清,其中包含p-cofilin蛋白成分。
1.2.4蛋白分步提取和Western印跡分析根據G-actin屬于胞質可溶性蛋白,F-actin為細胞骨架相關蛋白,采用不同緩沖液對這兩種蛋白進行分部提取。腦組織按每100 mg組織加入1 ml的組織蛋白裂解液(20 mmol·L-1Tris-Cl,pH 7.5,1 mmol·L-1Na3VO4,1%Triton X-100,5 mmol·L-1EGTA,1 mmol·L-1PMSF,1 mg·L-1aprotintin,1 mg·L-1pepstatin A,1 mg·L-1leupeptin),在 DY-89Ⅰ型電動勻漿器下勻漿30 s,冰浴30 min裂解后,4℃12 000 r·min-1離心30 min后取上清,即為G-actin的蛋白組分(含肌動蛋白單體G-actin)。沉淀按100 mg加入1 ml裂解液的比例重懸于緩沖液(10 mmol·L-1Tris-Cl,pH 7.5,150 mmol·L-1NaCl,1%Triton X-100,0.1%SDS,1 mmol·L-1脫氧膽酸鈉,2 mmol·L-1EDTA,1 mmol·L-1PMSF,1 mg·L-1aprotintin,1 mg·L-1pepstatin A,1 mg·L-1leupeptin)中冰浴30 min,間斷渦旋混勻,4℃ 12 000 r·min-1離心30 min后取上清,即為F-actin的蛋白組分[8]。BCA法蛋白定量,各取等量 G-actin組分及相應體積的F-actin組分,經SDS-PAGE電泳分離,電轉至PVDF膜上(4 ℃,2h),50 g·L-1脫脂奶粉的TBST封閉1 h,分別加入小鼠β-actin單克隆抗體(1∶500)、兔抗GAPDH多克隆抗體(1∶500),4℃過夜,TBST洗膜3次,每次10 min,然后加入相應辣根過氧化物酶標記的二抗(37℃,1h),DAB顯色。采用凝膠成像分析系統對Western blot區帶進行定量分析,P-cofilin蛋白的相對含量用其灰度值與GAPDH的灰度值比值來表示,肌動蛋白重構用F-actin/G-actin來表示。
1.2.5統計學分析采用SPSS 15.0軟件進行數據分析,Origin7.5軟件進行圖像處理。實驗數據以±s表示,采用單因素方差分析,Dunnett檢驗進行組間比較。
2 結果
2.1行為學觀察大鼠在注射納洛酮30 min后,嗎啡處理大鼠出現明顯的濕狗樣顫抖、吞咽、站立、跳躍、齒顫、上瞼下垂、流涎、腹瀉等戒斷癥狀,對戒斷癥狀進行評分,結果見Tab 1。與對照組相比,差異具有統計學意義(P<0.01),說明嗎啡依賴大鼠模型建立成功。
Tab 1 The withdrawal score in morphine dependent rats of different groups(±s)

Tab 1 The withdrawal score in morphine dependent rats of different groups(±s)
**P <0.01 vs control.C:control;M:morphine treatment
Group Withdrawal score 3.1 ±2.4 M 27.8 ±5.6 C**
2.2嗎啡依賴大鼠前額葉皮質中F-actin/G-actin、p-cofilin蛋白表達的變化與對照組相比,嗎啡依賴各組總actin的表達無變化,嗎啡依賴1周組大鼠前額葉皮質內F-actin/G-actin的比值沒有變化,嗎啡依賴2周組和4周組大鼠前額葉皮質內F-actin/G-actin的比值分別上調53%(P<0.05)和102%(P<0.01)。嗎啡依賴1周組大鼠前額葉皮質內pcofilin表達量沒有變化,嗎啡依賴2周組和4周組大鼠前額葉皮質內p-cofilin表達量分別上調93%(P<0.01)和88%(P<0.01)(Fig 1)。
2.3嗎啡依賴大鼠海馬中F-actin/G-actin、p-cofilin蛋白表達的變化與對照組相比,嗎啡依賴1周組和2周組大鼠海馬中F-actin/G-actin的比值沒有變化,嗎啡依賴4周組海馬中F-actin/G-actin比值上調94%(P<0.01)。嗎啡依賴1周組大鼠海馬中p-cofilin蛋白表達量沒有變化,嗎啡依賴2周組和4周組大鼠海馬中p-cofilin蛋白表達量分別上調45%(P<0.05)和30%(P<0.05)(Fig 2)。
2.4嗎啡依賴大鼠紋狀體中F-actin/G-actin、pcofilin蛋白表達的變化與對照組相比,嗎啡依賴各組大鼠紋狀體內F-actin/G-actin比值沒有變化。嗎啡依賴1周組大鼠紋狀體內p-cofilin蛋白表達量沒有變化,嗎啡依賴2周組和4周組的大鼠紋狀體內p-cofilin的蛋白表達量分別下調25%(P<0.05)和30%(P<0.05)(Fig 3)。
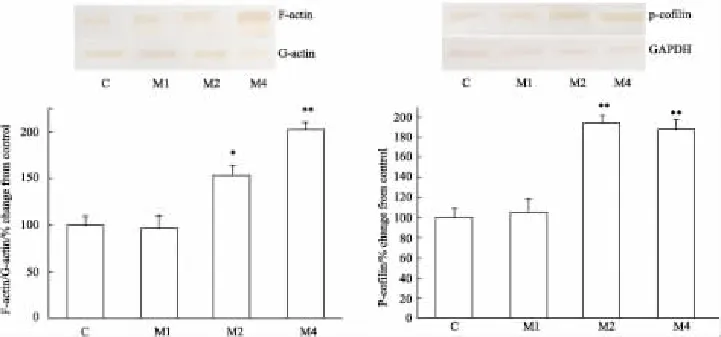
Fig 1 Expressions of F-actin/G-actin and p-cofilin in prefrontal cortex of morphine dependent rats(±s,n=5)

Fig 2 Expressions of F-actin/G-actin and p-cofilin in hippocampus of morphine dependent rats(±s,n=5)
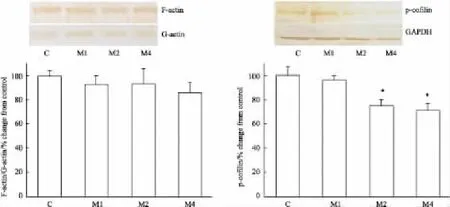
Fig 3 Expressions of F-actin/G-actin and p-cofilin in striatum of morphine dependent rats(±s,n=5)
2.5嗎啡依賴1、2和4周組大鼠丘腦中均未發現F-actin/G-actin、p-cofilin蛋白表達量發生明顯變化
3 討論
嗎啡濫用可造成神經系統突觸的可塑性改變,以往的研究表明[9-10],突觸可塑性包括很多環節,其中樹突棘的形態學改變被認為是促成各種刺激事件誘導的行為適應性改變的重要基礎,各種的刺激性事件就包括嗎啡成癮。這樣,嗎啡等精神藥物誘導的動物模型所表現出來的行為的可塑性與成癮性被聯系到樹突蛋白的持續性適應性改變,現在的研究認為樹突棘的形態學改變與其骨架肌動蛋白的重構有密切關系。肌動蛋白以單體和多聚體兩種表型存在,單體的肌動蛋白是由一條多肽鏈構成的球形分子,又稱球狀肌動蛋白(G-actin),肌動蛋白的多聚體形成肌動蛋白絲,稱為纖維狀肌動蛋白(F-actin)。肌動蛋白作為一種動態結構,持續進行組裝和解聚,而體內肌動蛋白的裝配及在細胞中的特性受肌動蛋白結合蛋白及上游信號分子的調節[3]。
Cofilin是中樞神經系統重要的肌動蛋白結合蛋白,Cofilin通過切斷F-actin,增加F-actin末端肌動蛋白亞單位的解離速度來調節其動力學[11]。Cofilin的活性依賴于它的磷酸化狀態,其受各種激酶及RhoGTPases的調節。RhoGTPase可通過 PAK、ROCK2/LIMK通路使cofilin磷酸化而降低其活性[12-13]。NMDA 受體激活后可以通過 NMDA/Ca2+/calcineurin/SSH通路使cofilin脫磷酸化而激活[14-15]。除了受磷酸化的調節之外,cofilin還與其它肌動蛋白結合蛋白發生相互作用,共同調節棘突的穩定性[16]。以上的研究提示,cofilin在樹突棘可塑性中可能發揮關鍵作用。然而p-cofilin在嗎啡依賴形成過程中的變化,國內外尚無報道。
本研究通過建立大鼠嗎啡依賴模型,選擇與嗎啡依賴關系密切的腦區:海馬、前額葉皮質、丘腦以及紋狀體,測定了嗎啡依賴狀態下F-actin、G-actin、p-cofilin在上述腦區的表達變化。研究結果顯示,在慢性嗎啡處理2周和4周大鼠的前額葉皮質以及嗎啡處理4周的海馬中F-actin/G-actin的比值均出現明顯上調。F-actin/G-actin比值的變化提示肌動蛋白骨架出現了重構,但是嗎啡依賴不同時程其變化不同,不同的腦區其變化亦不相同。
本研究顯示,肌動蛋白結合蛋白p-cofilin在嗎啡處理2周和4周的海馬和額葉皮質中均出現了表達上調,與F-actin的表達量增加相一致,提示嗎啡誘導的肌動蛋白重構可能與cofilin的磷酸化狀態有關。但p-cofilin在紋狀體中的表達卻減少了,提示不同的腦區可能存在著不同的調節機制。Cofilin的活性可影響神經元特異性肌動蛋白結合蛋白drebrin與F-actin的結合,進而影響棘突肌動蛋白網絡的穩定性[16],而drebrin的異常可影響突觸可塑性,導致機體不同程度的認知障礙[17]。行為學的研究表明cofilin與認知功能存在密切聯系[18],在阿爾采末病病人大腦的額皮質和海馬中,發現異常的cofilin-actin的棒狀結構。由于應激誘導的這種棒狀結構在軸突和樹突的形成,能夠導致神經性變性疾病[19]。在嗎啡成癮過程中均伴有不同程度的認知和行為障礙,提示p-cofilin嗎啡依賴誘導的肌動蛋白重構過程中可能發揮重要作用,但其上游信號調節過程還有待于進一步研究。
總之,我們的研究首次證明,大鼠嗎啡依賴時,伴隨著腦組織中肌動蛋白水平的變化,提示嗎啡依賴時大鼠腦組織中肌動蛋白發生了重構,肌動蛋白結合蛋白p-cofilin在嗎啡誘導的肌動蛋白重構中可能發揮重要作用。我們的結果對進一步闡明嗎啡成癮的細胞及分子生物學機制提供了實驗依據。
[1]Grewal S S,Fass D M,Yao H,et al.Calcium and cAMP signals differentially regulate cAMP-responsive element-binding protein function via a Rap1-extracellular signal-regulated kinase pathway[J].J Biol Chem,2000,275(44):34433 -41.
[2]Robinson T E,Kolb B.Structural plasticity associated with exposure to drugs of abuse[J].Neuropharmacology,2004,47(Suppl 1):33-46.
[3]Ethell I M,Pasquale E B.Molecular mechanisms of dendritic spine development and remodeling[J].Prog Neurobiol,2005,75(3):161-205.
[4]Pollard T D,Borisy G G.Cellular motility driven by assembly and disassembly of actin filaments[J].Cell,2003,112(4):453 -65.
[5]Toda S,Shen H W,Peters J,et al.Cocaine increases actin cycling:effects in the reinstatement model of drug seeking[J].J Neurosci,2006,26(5):1579 -87.
[6]Hou Y Y,Lu B,Li M,et al.Involvement of actin rearrangements within the amygdala and the dorsal hippocampus in aversive memories of drug withdrawal in acute morphine-dependent rats[J].J Neurosci,2009,29(39):12244 -54.
[7]Maldonado R,Koob G F.Destruction of the locus coeruleus decreases physical signs of opiate withdrawal[J].Brain Res,1993,605(1):128-38.
[8]溫進坤,史建紅,鄭 斌,等.SM22α對血管平滑肌細胞肌動蛋白聚合和交聯的調節[J].中國應用生理學雜志,2008,24(4):393-6.
[8]Wen J K,Shi J H,Zhang B,et al.The molecular mechanisms of SM22α in cytoskeleton remodeling of vascular smooth muscle cells[J].Chin Appl Physiol,2008,24(4):393 -6.
[9]Fukazawa Y,Saitoh Y,Ozawa F,et al.Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenancein vivo[J].Neuron,2003,38(3):447 -60.
[10]Yuste R,Bonhoeffer T.Morphological changes in dendritic spines associated with long-term synaptic plasticity[J].Annu Rev Neurosci,2001,24:1071 -89.
[11]DesMarais V,Ghosh M,Eddy R,Condeelis J.Confilin takes the lead[J].J Cell Sci,2005,118(pt 1):19 -26.
[12]Asrar S,Meng Y,Zhou Z,et al.Regulation of hippocampal longterm potentiation by p-21-activated protein kinase1(PAK1)[J].Neuropharmacology,2009,56(1):73 -80.
[13]Zhou Z,Meng Y,Asrar S,et al.A critical role of Rho-kinase ROCK2 in the regulation of spine and synaptic function[J].Neuropharmacology,2009,56(1):81 -9.
[14]Carlisle H J,Manzerra P,Marcora E,Kennedy M B.SynGAP regulates steady-state and activity-dependent phosphorylation of cofilin[J].J Neurosci,2008,28(50):13673 -83.
[15]Wang Y,Shibasaki F,Mizuno K.Calcium signal-induced cofilin dephosphorylation is mediated by Alingshot via calcineurin[J].J Biol Chem,2005,280(13):12683 -9.
[16]Kojima N,Shirao T.Synaptic dysfunction and disruption of postsynaptic drebrin-actin complex:a study of neurological disorders accompanied by cognitive deficits[J].Neurosci Res,2007,58(1):1-5.
[17]賈麗潔,羅 艷,張富軍,等.Drebrin對突觸可塑性的影響以及認知功能障礙的研究進展[J].中國藥理學通報,2010,26(8):989-92.
[17]Jia L J,Luo Y,Zhang F J,et al.Advance in research of the effects of Drebrin on synaptic plasticity and related cognitive dysfunction[J].Chin Pharmacol Bull,2010,26(8):989 -92.
[18]Rust M B,Gurniak C B,Renner M,et al.Learning,AMPA receptor mobility and synaptic plasticity depend on n-cofilin-mediated actin dynamics[J].EMBO J,2010,29(11):1889 -902.
[19]Peterson T S,Camden J M,Wang Y,et al.P2Y2 nucleotide receptor-mediated responses in brain cells[J].Mol Neurobio,2010,41(2-3):356-66.
猜你喜歡
作文周刊·小學二年級版(2022年20期)2022-05-05 01:33:06
娃娃樂園·綜合智能(2020年8期)2020-08-28 00:32:14
創新作文(小學版)(2019年10期)2019-09-25 08:12:28
作文周刊·小學二年級版(2018年9期)2018-04-18 10:01:40
小學生導刊(2018年1期)2018-03-15 08:02:37
小學生學習指導(低年級)(2017年5期)2017-05-04 04:14:38
科技知識動漫(2016年6期)2016-06-24 21:04:53
大灰狼(2015年6期)2015-07-16 21:01:00
作文與考試·小學高年級版(2015年17期)2015-05-30 10:48:04
汽車觀察(2009年1期)2009-02-18 09:11:50
