基于隱馬爾可夫模型的蛋白質序列篩選算法
2011-07-13 06:02:50張毅梅挺
電子設計工程 2011年21期
張毅,梅挺
(成都醫學院 人文信息管理學院,四川 成都 610083)
近年來,隨著人們對蛋白質測序工作的快速發展,蛋白質數據庫中的序列數量呈現指數級的增長速度,在這存儲有海量的蛋白質數據庫中,存在著大量的冗余蛋白質序列。雖然目前對冗余蛋白質序列尚未有很明確和統一的定義,但是普遍認為,在蛋白質數據庫中,如果兩條蛋白質序列的具有非常高的相似度,尤其是在整個序列中控制蛋白質功能的特征序列具有很強的相似性時,則認為這兩條蛋白質序列是互為冗余的序列。造成這種現象的原因很多,一個典型的原因即針對某一同源的蛋白質序列進行的測序,并將測量的結果存入數據庫中。
由于蛋白質數據庫在醫學研究、物種研究等方面發揮著非常重要的作用,利用蛋白質數據庫中的信息,有助于人們發現新物種,尋找物種之間的生物關系,研究針對某些特殊病毒的抗生藥物等。然而,如果在蛋白質數據庫中存在大量的冗余序列,則可能導致對這些蛋白質數據分析的誤差加大。比如在某一蛋白質序列簇中,如果冗余序列過多,可能會夸大這一序列簇的某些功能特征,從而對蛋白質序列間相互關系的研究產生誤導[1]。
目前,針對蛋白質數據庫冗余的問題,國內外有不少相關學者開展了研究,比較有代表性的有Hoblhm和Sander提出的CD-HIT去冗余算法,該算法的設計思想是在每個蛋白質序列簇中選取一個序列作為特征序列,然后再將該簇中的其余序列進行冗余檢測,如果某條序列與該序列的匹配程度超過某一閾值,則該序列被視為冗余序列。該算法是一種非常經典的蛋白質去冗余算法,目前也有很多算法是基于這一思想進行變形實現。此外還有在2000年由Yona和Linial共同提出了蛋白質序列聚類Protomap算法,Enright和Ouzounis提出了蛋白質序列分級聚類Generage算法,2004年由Kawaji和Takenaka提出了基于圖論的蛋白質序列分類算法。總體而言,這些蛋白質分類算法在分類的精確性和分類準確性兩方面還有待提高。
1 模型的建立與參數估計
1.1 模型的建立
從蛋白質序列數據庫中選擇一個蛋白質序列作為研究對象,記為D=D1D2…Dn,該研究對象也被稱為觀測對象。一個數據庫中的蛋白質序列也被視為隱馬爾可夫鏈的初始狀態的序列分布π,每一條蛋白質序列作為一個狀態的遷移中的中間狀態,觀測結果是初始狀態經過一定的遷移變化,且和一些隨機過程共同作用的狀態結果。狀態的遷移概率P1和觀測得到某一特定狀態結果的概率P2在分析之前需要進行確立,確立之后為隱馬爾可夫[2]可表示為 φ=(π,P1,P2)。
為了能夠對蛋白質數據庫中所有的序列進行匹配篩選,因此需要將蛋白質數據庫中的序列出現的概率進行抽象和描述。然后才能在此基礎上設定篩選的規則,并按照篩選規則確定的權值,篩選出最具有代表性的蛋白質序列。
假設在給定的隱馬爾可夫模型中,已經有觀測到某一序列的部分值為:{D1D2…Dk},且在時刻k,序列的狀態為 w1的概率[3]為:
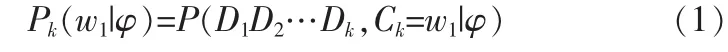
由于該概率的初始值可根據蛋白質數據庫中的常量計算得到,因此,該計算式可以采用遞歸的方式計算得到[4]:
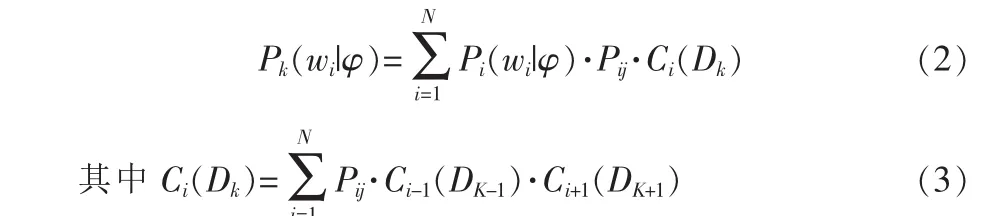
該式經過遞歸化簡后可得:
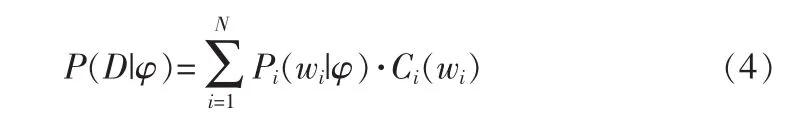
1.2 模型參數的估計
模型參數建立起來之后,將考慮如何選取合適的參數,使得對于一個給定的隱馬爾可夫模型φ=(π,P1,P2),如何得到其特定子序列的概率最大值,即P(D|φ)為最大。
假定對一個給定的隱馬爾可夫模型 φ=(π,P1,P2)和得到的觀測序列 D={D1,D2,…,Dn},在時刻 k 的狀態為 wi,時刻 k+1 的狀態為 wj,滿足這種特征的概率[5]記為 P(i,j),則有:
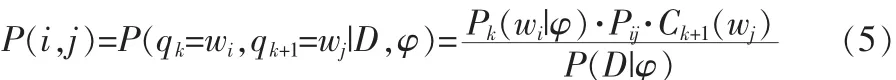
所以,通過上面的概率計算式,可以得到在模型確定的條件下,對于一個給定的觀測序列(即某一條蛋白質序列)下,k時刻的狀態為wi的概率[6]:
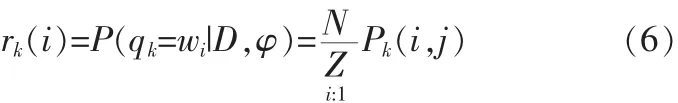
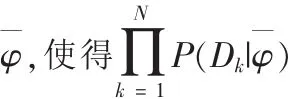
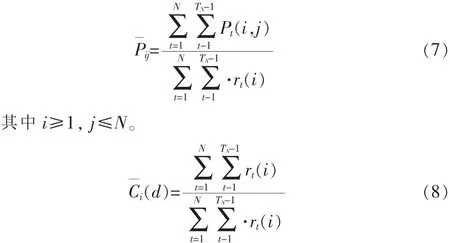
其中 N≥i≥1, N≥d≥1。
2 同源蛋白質序列篩選過程
蛋白質序列篩選過程中最為關鍵的是找出每個蛋白質序列的關鍵信息,這些信息直接決定了該蛋白質序列的主要功能和特征。因此定位和篩選出每條蛋白質序列中的關鍵信息是進行同源蛋白質序列篩選的重要前提。
蛋白質數據庫中每條序列記為D=D1D2-Dm-1Dm,隱馬爾可夫鏈階梯步長記為spl,在一條完整的序列中定義一個子片斷記為d=d1d2-dr-1dr。則子片斷d出現的概率[8]為:
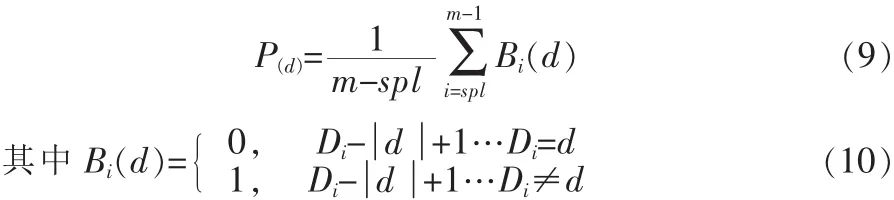
由此,可定義若存在子片斷d條件下,各個序列值的概率[9]:
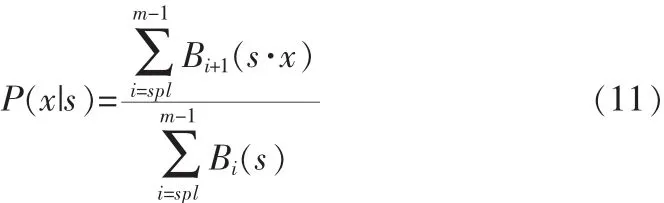
按照這種條件概率計算式,可以進一步得到子片斷d的詳細計算式[10]:
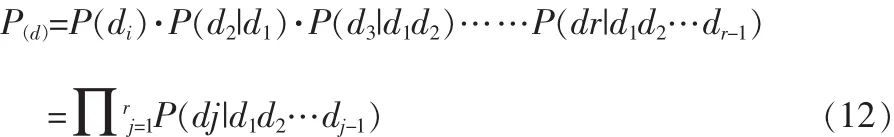
再定義整個蛋白質序列中,每一種可能的子片斷xd出現的概率[11]:
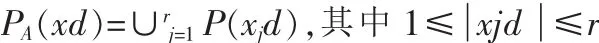
根據如上定義,可以得到任意一個子片斷的重要度描述參數Weight[12]。
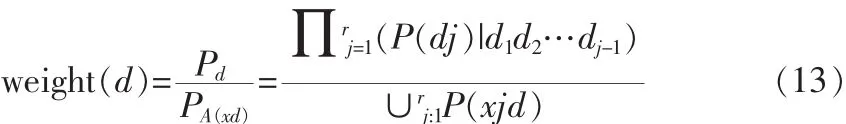
確定了任意一個子片斷的權值后,再按照序列的模式匹配,定位序列之間的匹配位置和匹配程度。若兩個對比的蛋白質序列長度不等,記 D1中有 d[1:x]特征序列,記 D2中有 d[1:y]特征序列。通過調整特征序列的長度,記錄兩個蛋白質序列中的最大匹配權值,該權值即為兩個蛋白質序列的匹配程度描述參數。對于兩個特征序列d[1:x]和d[1:y]的最大匹配權值計算式為[7]:
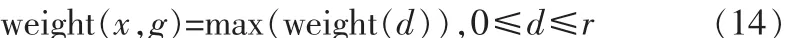
如此遞歸計算,最后得到最終結果。
3 測試結果
采用文中設計的基于隱馬爾可夫模型的蛋白質序列篩選算法SWISS-PROT對蛋白質數據庫中的數據進行了分類篩選測試,通過本文設計的算法,對蛋白質數據庫中的蛋白質序列特征進行提取和匹配,當匹配到兩條蛋白質序列的關鍵信息是一致的,則將這兩條蛋白質序列標記為同源蛋白質序列,同時將得到的篩選結果與目前公認的同源序列結果進行對比,得出篩選的正確率。測試結果如表1所示。

表1 基于隱馬爾可夫模型的蛋白質序列篩選算法測試結果Tab.1 Hidden Markov Model-based protein sequence selection algorithm results
測試結果表明,文中所設計的蛋白質篩選算法對蛋白質數據庫中篩選出了大量的同源蛋白質序列,根據與目前公認的同源蛋白質序列進行結果對比表明,在篩選過程中篩選出了蛋白質數據庫中的絕大多數的同源蛋白質序列,且篩選結果的正確率達到了95%以上,從篩選的精度而言,本設計的算法具有較高的篩選正確率。
4 結束語
從蛋白質數據庫中對蛋白質序列進行分類和篩選有著非常現實的意義,尤其是面對指數級增長的蛋白質數據庫,只有通過對蛋白質數據庫進行去冗余的處理,篩選出真正代表每個蛋白質簇的特征序列,才能建立更有實用價值的蛋白質數據庫。目前這一問題也是全球蛋白質序列研究的一個熱點問題,本文通過引入隱馬爾可夫模型對蛋白質序列進行分析篩選,探索了蛋白質數據庫特征序列篩選的新方法,已進行的測試結果也表明該方面在篩選的精度上可以達到95%以上。
[1]張成崗,歐陽曙光,張紹文,等.基于PC/Linux的核酸序列分析系統的構建及其應用 [J].生物化學與生物物理進展,2001(2):263-266.
ZHANG Cheng-gang, OU YANG Shu-guang, ZHANG Shaowen, et al.Based PC/Linux system, Construction and application of the nucleic acid sequence analysis system based on PC/Linux[J].Biochemistry and Biophysics,2001(2):263-266.
[2]陳英,彭心昭,樸英杰.自噬基因APG5基因結構的生物信息學分析[J].遺傳學報,2001,28(11):1077-1084.
CHEN Ying, PEN Xin-zhao, PU Ying-jie.Bioinformatics analysis of autophagy gene APG5 gene structure[J].Genetics,2001,28(11):1077-1084.
[3]齊建勛,肖奕.基于小波方法的蛋白質非規則二級結構預測[J].科學通報,2002(6):425-430.
QI Jian-xun,XIAO Yi.Non-wavelet-based method of protein secondary structure prediction rules[J].Chinese Science Bulletin,2002(6):425-430.
[4]任力鋒,張波,劉輝.蛋白質序列信息的提取與蛋白質結構預測[J].北京生物醫學工程,2005(3):237-238.
REN Li-feng, ZHANG Bo, LIU Hui.Protein sequence information extraction and protein structure prediction[J].Beijing Biomedical Engineering,2005(3):237-238.
[5]霍紅衛,肖智偉.基于最大權值路徑算法的DNA多序列比對方法[J].軟件學報,2007,18(2):185-195.
HUO Hong-wei,XIAO Zhi-wei.A multiplealignment approachforDNA sequencesbasedonthemaximum weighted path algorithms[J].Journal of Software,2007,18(2):185-195.
[6]鄒權,郭茂祖,王曉凱,等.基于關鍵字樹的DNA多序列星比對算法[J].電子學報,2009,37(8):1764-1850.
ZOU Quan, GUO Mao-zu, WANG Xiao-kai, etal.Keyword-based tree of the DNA sequence star more than the algorithm[J].Electronics Technology,2009,37(8):1764-1850.
[7]王艷春,何東健.神經網絡在蛋白質二級結構預測中的應用[J].安徽農業科學,2006(16):4172-4174.
WANG Yan-chun;HE Dong-jian,Neural network in protein secondary structure prediction in two[J].Anhui Agricultural Sciences,2006(16):4172-4174.
[8]阮曉鋼,孫海軍.編碼方式對蛋白質二級結構預測精度的影響[J].北京工業大學學報,2005,31(3):227-235.
RUAN Xiao-gang,SUN Hai-jun.Researchon encode influencing protein secondary structure prediction[J].Journal of Beijing University of Technology,2005,31(3):227-235.
[9]劉帥,馬志強,劉清雪,等.基于自適應免疫遺傳算法的多序列比對[J].信息技術,2007(2):15-17,111.
LIu Shuai, MA Zhi-qiang, LIU Qing-xue, et al.Adaptive immune genetic algorithm based on multiplesequence alignment[J].Information Technology,2007(2):15-17,111.
[10]郭衛斌,施保昌,王能超.多重生物序列對準及其算法綜述[J].高技術通訊,2001,11(6):96-102.
GUO Wei-bin, SHI Bao-chang, WANG Neng-chao.Multiple biological sequence alignment and its algorithm[J].High Technology,2001,11(6):96-102.
[11]關敏,辜華良,常雅萍,等.DNA核苷酸堿基序列分析軟件的編寫和應用[J],白求恩醫科大學學報,2001,27(5):467-469.
GUAN Min, GU Hua-liang, CHANG Ya-ping, et al.DNA nucleotide base sequence analysis software and application[J].BethuneUniversityofMedicalSciences,2001,27(5):467-469.
[12]杜世平.隱馬爾可夫模型在生物信息學中的應用[J].大學數學,2004,20(5):24-29.
DU Shi-ping. HMM in bioinformatics applications[J].University Mathematics,2004,20(5):24-29.
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
瘋狂英語·新策略(2019年10期)2019-12-13 08:43:28
當代陜西(2019年10期)2019-06-03 10:12:04
數學小靈通·3-4年級(2017年9期)2017-10-13 08:10:54
財經(2017年2期)2017-03-10 14:35:35
光學精密工程(2016年6期)2016-11-07 09:07:19
財經(2016年15期)2016-06-03 07:38:02
財經(2016年3期)2016-03-07 07:44:46
