頂空毛細管氣相色譜法測定長春西汀原料藥中乙酸乙酯殘留量
2011-08-07 05:58:46孫紅亞朱琦峰傅應華浙江寧波市鄞州人民醫院寧波市315040嘉興學院醫學院嘉興市314001
中國藥房 2011年45期
關鍵詞:檢測
孫紅亞,朱琦峰,傅應華#(1.浙江寧波市鄞州人民醫院,寧波市315040;.嘉興學院醫學院,嘉興市314001)
長春西汀是一種腦血管擴張藥,其原料藥質量標準——進口藥品注冊標準[1]中收載了乙酸乙酯殘留溶劑檢測項目,方法為以吡啶為溶劑、二氧六環為內標直接進樣測定,并規定了乙酸乙酯殘留溶劑限度為0.1%。筆者按該方法對長春西汀原料藥進行乙酸乙酯檢測時發現,因對照品溶液中乙酸乙酯濃度低,直接進樣存在檢測靈敏度低、峰面積小、重現性差、溶劑吡啶中雜質有干擾等問題,文獻也未見有關長春西汀殘留溶劑檢測方法報道。有關殘留溶劑測定有直接進樣法[2]和頂空進樣法[3,4]2種,為了提高檢測靈敏度和準確度,筆者根據《中國藥典》2010年版(二部)殘留溶劑測定法[5]的要求,對長春西汀中乙酸乙酯殘留溶劑檢測方法進行了改進研究:采用頂空進樣法,以二氧六環代替吡啶為溶劑,建立了以丙酮為內標的毛細管氣相色譜法測定長春西汀中乙酸乙酯殘留量,并對系統適用性進行了優化。結果表明方法簡單、靈敏、準確,可用于長春西汀原料藥中乙酸乙酯殘留量檢測。
1 儀器與試藥
6890N毛細管氣相色譜儀,包括氫火焰離子化檢測器(FID)、HP7694E頂空進樣器(美國Aglient公司);BP211D電子分析天平(北京賽多利斯儀器系統有限公司)。
長春西汀原料藥(6批均為進口。瑞士Linnea SA公司,批號:41809007、41809015、41809021,含量:99.5%、99.9%、99.2%;西班牙 Covex公司,批號:VP/VP070701、VP/VP/VP070901、CH080615GCH,含量:100.3%、99.5%、99.1%);乙酸乙酯、丙酮均為分析純,二氧六環為色譜純。
2 方法與結果
2.1 色譜條件與色譜
色譜柱:AgilentDB-624彈性石英毛細管柱(固定液為6%腈丙基苯基-94%聚二甲基硅氧烷,50m×0.535mm×3.00μm);柱溫:程序升溫,初始溫度55℃,保持6m in,并以40℃·m in-1速率升溫至180℃,保持2m in;載氣:高純氮氣,流速:1.0m L·m in-1;進樣口溫度:200 ℃;FID檢測口溫度:250 ℃;進樣方式:分流進樣,分流比:5∶1;頂空溫度:85℃;平衡時間:30 m in;進樣時間:1m in。
取“2.2”項下混合對照品溶液、二氧六環、供試品溶液進樣分析,結果乙酸乙酯與內標丙酮峰分離良好,分離度大于1.5。色譜見圖1。
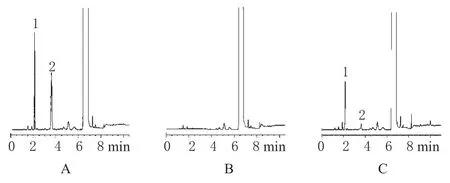
圖1 氣相色譜圖A.混合對照品溶液;B.二氧六環;C.供試品溶液;1.內標;2.乙酸乙酯Fig 1 GC chromatogramsA.control solution mixture;B.dioxane;C.sample solution;1.internal standard;2.ethylacetate
2.2 溶液制備及測定方法
精密稱取丙酮適量,用二氧六環稀釋制成0.2mg·m L-1的溶液,作為內標貯備液;精密量取貯備液5m L,置于50m L容量瓶中,加二氧六環至刻度,作為內標溶液。
精密稱取乙酸乙酯50.06mg,置于100m L容量瓶中,加二氧六環至刻度,作為對照品貯備液。精密量取對照品貯備液與內標貯備液各5m L,置于50m L容量瓶中,加二氧六環至刻度,搖勻;再精密量取10m L,置于20m L頂空瓶中,加蓋密封,作為混合對照品溶液。
取供試品約0.5 g,置于20m L頂空瓶中,精密稱定;再精密加入內標溶液10m L,加蓋密封,振搖使溶解,作為供試品溶液。
取混合對照品溶液與供試品溶液,于85℃加熱30m in,照《中國藥典》2010年版(二部)附錄殘留溶劑測定法[5],以“2.1”項下色譜條件測定,按內標法以峰面積計算。
2.3 線性范圍
精密量取“2.2”項下對照品貯備液0.5、1.0、2.0、3.0、5.0 m L,分別置于20m L頂空瓶中,各精密加入內標貯備液1m L,分別加二氧六環至10m L,搖勻,加蓋密封,依法測定。以乙酸乙酯峰面積與內標峰面積之比(Y)為縱坐標,濃度(X,mg·m L-1)為橫坐標進行線性回歸,得乙酸乙酯的回歸方程為:Y=10.345X+0.040 2(r=0.999 8)。表明乙酸乙酯檢測濃度線性范圍為0.025~0.25mg·m L-1。
2.4 精密度試驗
取“2.2”項下混合對照品溶液,重復測定6次,計算乙酸乙酯峰面積與內標峰面積的平均比值為1.086,RSD=1.08%(n=6),表明精密度良好。
2.5 溶液穩定性試驗
取“2.2”項下混合對照品溶液,分別于0、2、4、6、8 h時各取樣測定,得乙酸乙酯峰面積與內標峰面積比值無明顯差別,表明溶液在8 h內穩定。
2.6 最低定量限試驗
取“2.2”項下混合對照品溶液,用二氧六環逐級稀釋成不同濃度,依法測定,按S/N=10計算最低定量濃度為0.10μg·m L-1。
2.7 回收率試驗
取經105℃干燥至恒重的某批樣品約0.5 g,共6份,置于20m L頂空瓶中,精密稱定,分別精密加入“2.2”項下混合對照品溶液10m L,加蓋密封,振搖使溶解,按“2.2”項下方法依法測定,按內標法以峰面積計算回收率,得平均回收率為102.2%,RSD=1.82%(n=6),詳見表1。
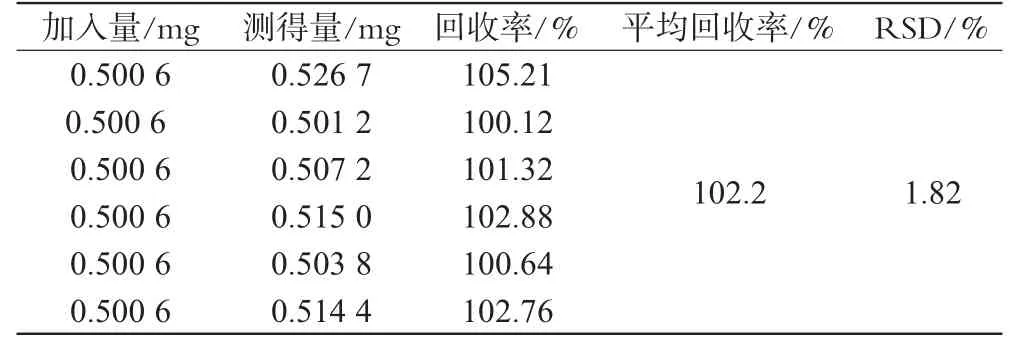
表1 回收率試驗結果(n=6)Tab 1 Resultsof recovery tests(n=6)
2.8 樣品中乙酸乙酯殘留量測定
精密稱取6批長春西汀原料藥各0.5 g,按照“2.2”項下方法制備混合對照品溶液和供試品溶液,依法測定,結果批號為41809021的樣品中檢出乙酸乙酯殘留量為0.023%,其余5批均未檢出;同時取樣按照進口藥品注冊標準方法[1]檢驗,結果6批樣品均未檢出乙酸乙酯。
3 討論
3.1 溶劑的選擇
長春西汀易溶于氯仿,在二甲基甲酰胺中略溶。考慮到氯仿毒性大,不宜作為溶劑使用。在前期研究中經試驗二甲基甲酰胺、二甲基亞砜和二氧六環3種溶劑,發現二甲基甲酰胺、二甲基亞砜對長春西汀的溶解度均不能滿足樣品濃度的要求,而二氧六環對照品的溶解度能達到5%的要求,故選擇二氧六環作溶劑。
3.2 內標物與色譜柱的選擇
筆者曾采用丙酮、乙醇、正丙醇作為內標,經試驗三者均能與乙酸乙酯分離,但正丙醇在二氧六環溶劑中存在峰形變寬問題,柱效降低,不適合作內標;長春西汀化學結構中含有乙醇酯基團,經考察在90℃下加熱30min頂空進樣,樣品在乙醇峰位處出現1個色譜峰,經比較確認為長春西汀受熱分解產生的乙醇峰,故乙醇不適合作內標;丙酮的保留時間較為適宜,在給定的色譜條件下,與乙酸乙酯分離良好,其峰形不受溶劑影響,柱效高,故選擇丙酮作為內標。
筆者曾考察了HP-5、HP-INNOWax和DB-624 3種色譜柱的適用性,結果表明以DB-624色譜柱效果最好,乙酸乙酯、乙醇、內標、溶劑峰分離良好,乙酸乙酯、內標峰形對稱、柱效高、檢測靈敏度高、重現性好,故選擇DB-624為色譜柱。
3.3 頂空平衡溫度和時間選擇等
取混合對照品溶液3份,分別在70、80、85℃下加熱平衡30m in后進樣測定。結果表明,乙酸乙酯和內標的峰面積隨平衡溫度升高而增大。
另取混合對照品溶液2份,分別在85℃加熱30、45min后進樣測定,結果在平衡30、45min時測得乙酸乙酯和內標的峰面積基本不變。故選擇加熱溫度(即頂空溫度)為85℃、平衡時間為30min。
采用頂空進樣法,可大大提高乙酸乙酯的檢測靈敏度,同時還可減少樣品對色譜柱的污染,延長柱子的使用壽命。采用程序升溫,先在低溫下完成乙酸乙酯和內標的分離,再提高柱溫,使高沸點溶劑二氧六環快速出峰,達到快速分析的目的。
進口藥品注冊標準[1]中規定長春西汀中乙酸乙酯限量為0.1%,較《中國藥典》2010年版(二部)規定的乙酸乙酯殘留量限度0.5%低了5倍,因此需要更高的檢測靈敏度。而本法具有檢測靈敏度更高、重復性和準確度更好的優點,可用于長春西汀原料藥中乙酸乙酯殘留量的檢測,也可為長春西汀質量標準的修訂提供方法依據。
[1]國家食品藥品監督管理局.進口藥品注冊標準——長春西汀,JX20070232[S].2009.
[2]張雅軍,孫會敏,李 科,等.氣相色譜法測定阿德福韋酯中的10種殘留溶劑[J].藥物分析雜志,2005,25(6):660.
[3]趙佳麗,章展煌,徐宏祥.頂空毛細管氣相色譜內標法測定倍他米松磷酸鈉中的3種殘留溶劑[J].中國藥房,2010,21(21):1 999.
[4]席志芳,衷紅梅,黨全訓,等.頂空氣相色譜法測定泮托拉唑鈉中殘留的有機溶劑[J].藥物分析雜志,2007,27(7):1 119.
[5]國家藥典委員會.中華人民共和國藥典(二部)[S].2010年版.北京:中國醫藥科技出版社,2010:80、附錄ⅧP.
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
