不同成因類型黃銅礦細菌浸出鈍化
2011-08-09 01:00:48傅開彬林海莫曉蘭董穎博汪涵
中南大學學報(自然科學版) 2011年11期
傅開彬,林海,莫曉蘭,董穎博,汪涵
(北京科技大學 土木與環境工程學院,北京,100083)
黃銅礦屬于原生礦,儲量豐富,就化學和細菌浸出而言,是最難處理的硫化銅礦物[1]。常溫下細菌浸出,速率慢,浸出率低,一般在20%~30%之間[2]。通常認為鈍化是黃銅礦浸出率低的原因,在細菌浸出過程中,所謂鈍化是指礦物表面形成阻礙層,限制了細菌、營養物質、氧化劑與礦物接觸,反應產物難以離開礦物表面,從而抑制浸出繼續進行的現象[3]。目前,許多研究者提出了鈍化黃銅礦生物浸出的幾種可能阻礙層,有黃鉀鐵礬層、硫層、中間硫化產物層(多硫化物)和氫氧化鐵層等,其中以前3種觀點最為普遍。硫阻礙論者[4?6]認為在礦物表面形成的元素硫及其多聚物阻止了黃銅礦浸出;黃鉀鐵礬論者[7?9]認為含鐵羥基化物,特別是覆蓋在礦物表面的黃鉀鐵礬才是導致黃銅礦浸出率低的原因,其沉淀直接減少了具有氧化作用的Fe3+,使其濃度降低;多硫化物論者[10?12]認為,黃銅礦浸出率的下降,是由于礦物表面銅鐵擴散速率不同,鐵優先浸出,形成金屬缺陷的多硫化物層阻礙了Cu2+和Fe2+的傳輸,同時也減緩了反應電子的傳遞。對鈍化層認識不一致,與影響黃銅礦生物浸出的因素多、關系復雜有關,也與礦物成因、浸出條件和研究手段等有關[13]。黃銅礦成因類型與其在細菌浸出過程中的鈍化形式之間關系的研究較少。本文作者利用自主分離的嗜酸氧化亞鐵流桿菌 LD-1為浸礦菌種,采用X線衍射技術(XRD)、掃描電鏡(SEM)和X線光電子能譜(XPS)等技術手段,研究了不同成因類型黃銅礦(斑巖型、黃鐵礦型)的細菌浸出及其鈍化現象,以了解黃銅礦成因類型與鈍化形式之間的聯系。
1 原料及研究方法
1.1 原料
試驗用的純礦物購自浙江大學地質標本廠,黃鐵礦型黃銅礦源自浙江紹興漓渚鐵礦,斑巖型黃銅礦來自江西德興銅礦。將礦石清洗脫泥,自然晾干,無污染條件下進行破碎,然后手選挑純。挑選出來的純礦物在瓷球磨機內濕磨至粒度小于0.074 mm,過濾,冷凍干燥后,磨口瓶保存備用,送XPS檢測的樣品采用氮氣封存。
對樣品進行 X線衍射和化學成分分析(質量分數)。結果表明:黃鐵礦型黃銅礦主要含有黃銅礦,以及少量黃鐵礦,銅品位為27.38 %,鐵品位為28.35%,硫品位為33.31%;斑巖型黃銅礦主要含有黃銅礦,以及少量黃鐵礦和石英,銅品位為 27.88%,鐵品位為28.29%,硫品位為32.36%。依據化學分析結果計算純度,黃鐵礦型黃銅礦為 79.22%,斑巖型黃銅礦為80.62%。
1.2 菌種和培養基
試驗菌種嗜酸氧化亞鐵流桿菌 LD-1采自湖北某礦山,經過篩選、培養、馴化和分離得到,菌種最佳培養條件為:搖床轉速160 r/min,溫度30 ℃,pH 2,采用改進的4.5 K培養基,以硫酸亞鐵為能源物質傳代培養,其配方如下:(NH4)2SO42.0 g,KCl 0.1 g,K2HPO40.25 g,MgSO4·7H2O 0.25 g,Ca(NO3)20.01 g,FeSO4·7H2O 22.2 g,H2O 1 000 mL。用 10%的稀硫酸調節培養基pH。
1.3 細菌浸出試驗
浸出試驗采用250 mL三角瓶為容器,瓶內裝入100 mL溶液,礦漿濃度為20 g/L,接種量為6×108個。用pH為2.0稀硫酸預處理,10%的稀硫酸溶液調節pH至2.0,待pH穩定,接入馴化后對數生長期的菌LD-1。接種后,在恒溫空氣振蕩培養箱中進行培養,轉速為160 r/min,溫度為30 ℃。每隔4 d取樣1 mL,測試浸出液中Cu2+的質量濃度,取樣前用蒸餾水補足蒸發掉的水分,測試消耗的液量用相應的溶液補充,保證溶浸液總體積不變。
浸渣用pH為2的稀硫酸反復調漿清洗,最后用去離子水于3 000 r/min離心處理,冷凍干燥,氮氣封存后立即送檢測。
1.4 測試分析方法
用碘量法測定浸出液中Cu2+質量濃度;細菌用血球計數板在顯微鏡下直接計數;用X線衍射技術(XRD)分析浸出前、后黃銅礦的礦物組成;用掃描電鏡(SEM)觀察礦物表面的浸蝕特征;用 X線光電子能譜(XPS)分析黃銅礦表面層物質組成。
2 結果及討論
2.1 銅浸出速率變化
利用軟件 origin8將浸出液中 Cu2+質量濃度對時間求導,得到Cu2+浸出速率變化如圖1所示。由圖1可見:2種黃銅礦浸出速率變化趨勢相似,都是先升高,后降低,最后接近于0,但相差較大。試驗初期,浸出速率都很慢,但斑巖型黃銅礦浸出速率比黃鐵礦型黃銅礦浸出速率大,且增長迅速,浸出14 d時,先達到最大值,為6.3×10?2g/(L·d),在其浸出速率開始降低時,黃鐵礦型黃銅礦浸出速率開始增長,到第28 d時,上升達到最大,為27.3×10?2g/(L·d),試驗后期,浸出速率都逐漸接近于0。
在細菌浸出過程中,黃銅礦浸出速率下降是由于在礦物表面逐漸形成了鈍化層[14],但兩類黃銅礦表面形成鈍化層的性質不同,斑巖型黃銅礦表面鈍化層比黃鐵礦型黃銅礦表面鈍化層形成速率更快,且對浸出的阻礙能力更強。
2.2 浸渣XRD分析
2種不同成因類型黃銅礦浸渣XRD分析結果如圖2和3所示。從圖2可以看出:浸渣中主要含有黃銅礦、硫的多聚物S8、硫砷銅礦和黃鐵礦等,與原礦相比出現硫多聚物。圖3結果表明:浸渣中主要含有黃銅礦、硫化銅鐵(Cu18.32Fe15.9S32)、硫砷銅礦和輝銅礦(Cu2S)等,與原礦相比黃鐵礦基本浸蝕完全,新增硫化銅鐵和輝銅礦。XRD分析結果表明:2種不同成因類型的黃銅礦浸出效果不同,浸渣成分也不一樣,其反應途徑應該有所區別。
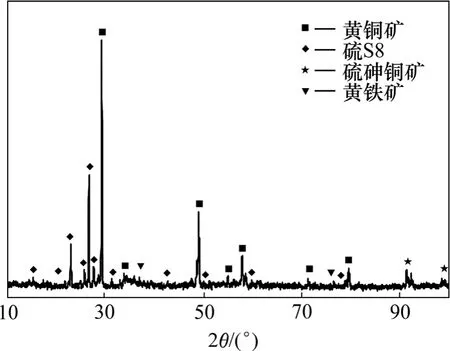
圖2 黃鐵礦型黃銅礦浸渣的XRD譜Fig.2 XRD pattern of pyritic chalcopyrite leaching residue

圖3 斑巖型黃銅礦浸渣的XRD譜Fig.3 XRD pattern of porphyry chalcopyrite leaching residue
張在海等[15]研究 CuFeS2細菌氧化中的礦物轉化過程,認為其細菌氧化可能按如下順序先后產生中 間 產 物 : CuFeS2→Cu9Fe9S16→CuFeS1?x→CuS2→Cu9S5→Cu2S→CuS→Cu2O(CuCl,CuCl2,Cu2OCl2)→Cu3SO4(OH)4→CuSO4。Scott[16]將輝銅礦制成電極,研究了輝銅礦的細菌氧化過程,測定由Cu2S到CuS的中間反應,它們反應順序如下:Cu2S→Cu1.97S→Cu1.8S→Cu1.6S→Cu1.4S→Cu1.12S→CuS。這些中間反應極快地完成,如Cu2S到Cu1.8S僅用3.5 min就完全轉化。因此,CuFeS2→Cu2S是黃銅礦細菌浸出的關鍵,該階段銅鐵比例為1:1,銅鐵的溶解速率相同。而斑巖型黃銅礦浸渣中出現了硫化銅鐵Cu18.32Fe15.9S32,則說明其礦物轉化過程并沒有完全按照上述模型進行,礦物表面銅、鐵離子浸出速率有差異,鐵離子遷移速率較快。
2.3 浸渣SEM-EDS分析
浸渣表面的掃描電鏡照片和能譜分析結果如圖4和5所示。從圖4可以看出:黃鐵礦型黃銅礦浸渣表面浸蝕現象明顯,有顆粒狀物質,結構疏松,干燥后出現裂紋,能譜分析表明,硫峰很高,銅和鐵峰較低,結合 XRD分析結果,表面應該是硫或者硫的多聚物。由圖5可見:斑巖型黃銅礦浸渣顆粒表面光滑致密,有少量的浸蝕坑。從圖5(b)可以看出:與原礦相比,表面銅、鐵和硫的峰都有所降低,而相對含量發生了改變,銅含量上升,鐵含量下降。通過分析可以看出:斑巖型黃銅礦浸渣表面致密結構的鈍化能力強于黃鐵礦型黃銅礦表面以硫為主的疏松阻礙層的鈍化能力。

圖4 黃鐵礦型黃銅礦浸渣SEM-EDS結果Fig.4 SEM-EDS results of pyritic chalcopyrite leaching residue

圖5 斑巖型黃銅礦浸渣SEM-EDS結果Fig.5 SEM-EDS results of porphyry chalcopyrite leaching residue
2.4 浸渣XPS分析
為了進一步確認黃銅礦浸渣表面阻礙層的物質組成,利用X線光電子能譜分析原礦和浸渣表面銅、鐵和硫的結合能和含量變化。
采用英國Kratos公司生產的AXIS Ultra DLD型XPS譜儀。選擇Al Kα靶作為激發源,電壓為15 kV,功率為250 W。譜峰均以樣品中的C 1s峰(Eb=284.6 eV)為參考進行校正,以消除荷電效應的影響。
2.4.1 黃鐵礦型黃銅礦XPS分析
黃鐵礦型黃銅礦原礦和浸渣表面銅、鐵和硫的X線光電子能譜分析結果如圖6所示。從圖6可以看出:黃鐵礦型黃銅礦浸渣表面銅、鐵和硫的含量與原礦相比發生了較大的變化,浸渣表面銅峰完全消失,鐵和硫峰強度降低。浸出前后礦物表面銅、鐵和硫含量見表1。
表1中數據表明:黃鐵礦型黃銅礦浸渣表面銅的相對含量為0,鐵含量較低,為19.74%,以硫為主,為80.26%。圖6(c)所示為S 2p譜。由圖6(c)可見:與細菌作用后,礦物表面S 2p峰從161.8~162.7 eV上升到163.7 eV,主要是因為原礦中含有黃銅礦(162.7 eV)和少量黃鐵礦(161.8 eV),但雙峰不明顯,低價態的硫被氧化為硫單質或多聚物(163.7 eV)沉淀在礦物表面,所以,浸渣表面出現S0峰。
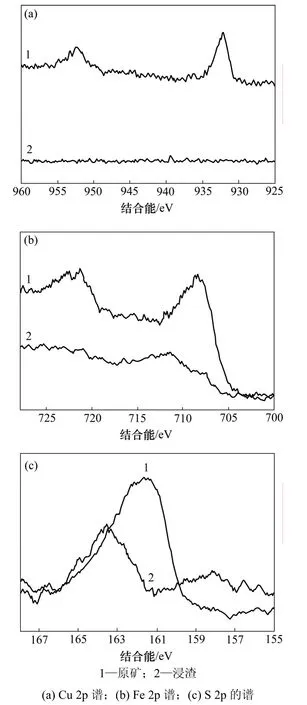
圖6 浸出前和浸出后黃鐵礦型黃銅礦表面的XPS譜Fig.6 XPS spectra of pyritic chalcopyrite before and after bio-leaching

表1 浸出前、后黃鐵礦型黃銅礦表面銅、鐵和硫含量(質量分數)Table1 Content of Cu, Fe and S on surface of pyritic chalcopyrite before and after bio-leaching %
圖6(a)所示為黃鐵礦型黃銅礦表面Cu 2p譜。原礦表面Cu的強峰在932.4 eV處,表示3d軌道沒有完全充滿,處于激發態,這與純的Cu(Ⅰ)不匹配,Todd等[17]通過黃銅礦、銅藍和輝銅礦表面銅結合能的比較,認為黃銅礦中銅離子應為+2價,結構式為:Cu2+Fe2+S2,鐵 L-邊光譜分析結果也證實了這一結構的正確性,浸渣表面銅被單質硫或多聚物覆蓋,沒有檢測到Cu 2p峰。原礦表面Fe(Ⅱ)(708.9 eV)(見圖6(b))被氧化為Fe(Ⅲ)(711.8 eV),少量吸附或夾雜在硫中。
綜上所述,黃鐵礦型黃銅礦浸渣表面以硫單質為主,吸附少量Fe3+,其表面阻礙層應為硫及其多聚物。
2.4.2 斑巖型黃銅礦XPS分析
斑巖型黃銅礦X線光電子能譜分析結果如圖7所示。從圖7可以看出:浸出前和浸出后斑巖型黃銅礦表面銅、鐵和硫相對含量發生了變化,銅峰向低漂移,鐵峰向高處移動,硫峰強度增大,峰位基本不變。3種元素含量見表2。
從表2中可以看出:與原礦相比,浸渣中銅的相對含量增加至27.0%,鐵減少為 13.13%,硫含量增加為59.87%。
從圖7(a)可知:浸渣表面銅峰強度升高,原礦表面Cu(Ⅱ)的結合能為933.2 eV,細菌浸出后,表面形成Cu(Ⅰ),而結合能降低為932.2 eV,結合能變化規律符合 Hiroyoshi等[18]提出黃銅礦浸出的 2步溶解模型。
圖7(b)所示為斑巖型黃銅礦表面Fe 2p譜。從圖7(b)可知:浸渣表面鐵的強度降低,結合能向高偏移,價態發生了改變,原礦表面Fe(Ⅱ)(708.8 eV)被氧化成 Fe(Ⅲ)(711.8 eV),使 Fe(Ⅲ)峰升高,Fe(Ⅱ)峰強度降低。
然而,細菌浸出后,硫結合能(見圖7(c))沒有變化,只是強度升高,S 2p雙峰更為明顯,主要是因為浸渣表面存在黃銅礦(162.7 eV)和輝銅礦Cu2S(161.8 eV)。
通過以上分析,可以看出浸渣表面含有Cu+,Fe3+和S2?,由這些離子組成的具有特殊結構的富銅貧鐵層阻礙了斑巖型黃銅礦的浸出。
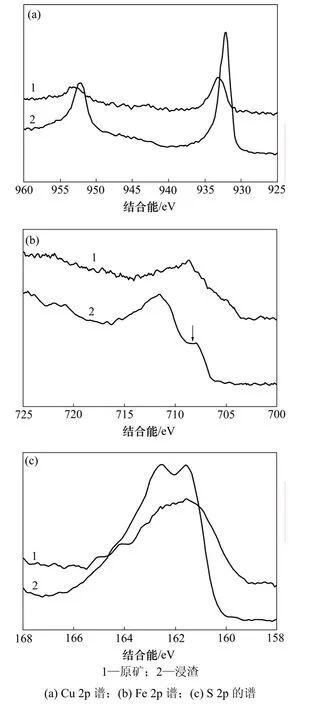
圖7 浸出前和浸出后斑巖型黃銅礦表面XPS譜Fig.7 XPS spectra of porphyry chalcopyrite before and after bio-leaching
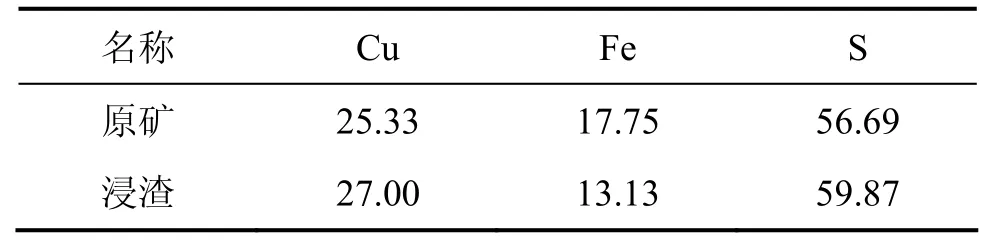
表2 浸出前和浸出后斑巖型黃銅礦表面銅、鐵和硫含量(質量分數)Table2 Content of Cu, Fe and S on surface of porphyry chalcopyrite before and after bio-leaching %
3 結論
(1)2種黃銅礦浸出速率變化規律相似,都是先升高,后降低,最后接近于0,但差異較大。試驗初期,斑巖型黃銅礦浸出速率比黃鐵礦型黃銅礦浸出速率大,且增長迅速,14 d時,先達到最大值 6.3×10?2g/(L·d),在其浸出速率開始降低時,黃鐵礦型黃銅礦浸出速率開始增長,到28 d時,達到最大值,為27.3×10?2g/(L·d)。
(2)在細菌浸出過程中,黃銅礦浸出速率下降是由于在礦物表面逐漸形成了鈍化層,但兩類黃銅礦表面形成鈍化層的性質不同,斑巖型黃銅礦表面鈍化層比黃鐵礦型黃銅礦鈍化層形成速度更快,且對浸出的阻礙能力更強。
(3)黃鐵礦型黃銅礦浸渣中主要含有黃銅礦、硫多聚物S8、硫砷銅礦和黃鐵礦等,與原礦相比,出現硫多聚物。斑巖型黃銅礦浸渣中主要含有黃銅礦、硫化銅鐵Cu18.32Fe15.9S32、硫砷銅礦和輝銅礦等,黃鐵礦被完全浸蝕,產生了硫化銅鐵和輝銅礦等。
(4)黃鐵礦型黃銅礦浸渣表面浸蝕現象明顯,結構疏松;斑巖型黃銅礦浸渣顆粒表面光滑致密,有少量浸蝕坑。
(5)通過X線光電子能譜可以確定,黃鐵礦型黃銅礦浸渣表面阻礙層應為硫及其多聚物;而斑巖型黃銅礦浸渣表面含有Cu2+,Fe3+和S2?,由這些離子組成的具有特殊結構的富銅貧鐵層阻礙了浸出反應繼續進行。
[1]Arce E M, Gonzalez I.A comparative study of electrochemical behavior of chalcopyrite, chalcocite and bornite in sulphuric acid solution[J].International Journal of Mineral Processing, 2002,67(1/2/3/4): 17?28.
[2]Rodriques Y, Ballester A, Blazques M L, et al.New information on the pyrite bioleaching mechanism at low and high temperature[J].Hydrometallurgy, 2003, 71(1/2): 37?46.
[3]Stott M B, Watling H R, Franzmann P D, et al.The role of iron-hydroxy precipitates in the passivation of chalcopyrite during bioleaching[J].Minerals Engineering, 2000, 13(10):1117?1127.
[4]Dutrizac J E.Elemental sulphur formation during the ferric sulphate leaching of chalcopyrit[J].Canadian Metallurgical Quarterly, 1989, 28(4): 337?344.
[5]Klauber C, Parker A, Bronswijk W V, et al.Sulphur speciation of leached chalcopyrite surfaces as determined by X-ray photoelectron spectroscopy[J].International Journal of Mineral Processing, 2001, 62(1/4): 65?94.
[6]Bevilaqua D, Diezperez I, Fugivara C S, et al.Oxidative dissolution of chalcopyrite by Acidithiobacillus ferrooxidans analyzed by electrochemical impedance spectroscopy and atomic force microscopy[J].Bioelectrochemistry, 2004, 64(1): 79?84.
[7]Sandstrom A, Shchukarev A, Paul J.XPS characterisation of chalcopyrite chemically and bioleached at high and low redox potential[J].Minerals Engineering, 2005, 18(5): 505?515.
[8]Third K A, Cordruwisch R, Watling H R.The role of iron-oxidizing bacteria in stimulation or inhibition of chalcopyrite bioleaching[J].Hydrometallurgy, 2000, 57(3):225?233.
[9]Parker A, Klauber C, Kougianos A, et al.An X-ray photoelectron spectroscopy study of the mechanism of oxidative dissolution of chalcopyrite[J].Hydrometallurgy, 2003, 71(1/2): 265?276.
[10]Parker A J, Paul R L, Power G P.Electrochemistry of the oxidative leaching of copper from chalcopyrite[J].Journal of Electroanalytical Chemistry, 1981, 118: 305?316.
[11]Acero P, Cama J, Ayora C.Kinetics of chalcopyrite dissolution at pH 3[J].European Journal of Mineralogy, 2007, 19(2): 173?182.
[12]Lazaro I, Nicol M J.The mechanism of the dissolution and passivation of chalcopyrite:an electrochemical study[C]//Young C, Alfantazi A M, Anderson C G, et al.Hydrometallurgy 2003.Warrendale, PA: The Minerals, Metals & Materials Society, 2003:405?417.
[13]舒榮波, 阮仁滿, 溫建康.黃銅礦生物浸出中鈍化現象研究進展[J].稀有金屬, 2006, 30(3): 395?400.SHU Rong-bo, RUAN Ren-man, WEN Jian-kang.Review on passivation of chalcopyrite during bioleaching process[J].Chinese Journal of Rare Metals, 2006, 30(3): 395?400.
[14]Klauber C.A critical review of the surface chemistry of acidic ferric sulphate dissolution of chalcopyrite with regards to hindered dissolution[J].International Journal of Mineral Processing, 2008, 86(1/2/3/4): 1?17.
[15]張在海, 王淀佐, 鄧吉牛, 等.黃銅礦細菌轉化與浸出機理探討[J].中國工程學, 2005, 7(增刊): 266?268.ZHANG Zai-hai, WANG Dian-zuo, DENG Ji-niu, et al.Discuss on the bacterial transformation and leaching mechanism of chalcopyrite[J].Engineering Science, 2005, 7(S): 266?268.
[16]Scott D J.The mineralogy of copper leaching: Concentrates and heaps, copper’95[C]//Copper Hydrometallurgy Short Course.Santiago, 1995: 65.
[17]Todd E C, Sherman D M, Purton J A.Surface oxidation of chalcopyrite (CuFeS2)under ambient atmospheric and aqueous(pH 2-10)conditions: Cu, Fe L- and O K-edge X-ray spectroscopy[J].Geochimica et Cosmochimica Acta, 2003,67(12): 2137?2146.
[18]Hiroyoshi N, Miki H, Hirajima T, et al.A model for ferrous-promoted chalcopyrite leaching[J].Hydrometallurgy,2000, 57(1): 31?38.
