密度泛函計算中基組的收斂現象:強成鍵體系CO分子
2011-12-08 06:01:32黃鶯雷志丹張曉青李龍
湖南師范大學自然科學學報 2011年5期
關鍵詞:性質
黃鶯,雷志丹,張曉青,李龍
(湖南中醫藥大學藥學院,中國長沙 410208)
密度泛函計算中基組的收斂現象:強成鍵體系CO分子
黃鶯*,雷志丹,張曉青,李龍
(湖南中醫藥大學藥學院,中國長沙 410208)
探討了用密度泛函理論中7個常見交換-相關密度泛函及8種常見Dunning調和相關基組處理CO分子的三種性質(平衡鍵長、諧振基頻和離解能)時表現出的基組收斂現象.用Dunning的單指數插補法,求得了上述性質的全基組極限值.用擴展的調和相關基組,發現鍵長的系統誤差在0.002 9(LDA)到0.007 3 ?(BP86)之間,基頻誤差為17(LDA)至56(B3BP86)cm-1,離解能誤差為12.6(BLYP)至186.6(LDA)kJ/mol.這些結果對系統地認識波函數的基組收斂現象將提供有益啟示.
密度泛函理論;基組收斂;調和相關基組
近年來,以密度泛函理論(DFT)為基礎的計算方法的運用得到了廣泛的普及和承認[1-6].DFT的理論基礎源于Hohenberg和Kohn的兩個著名定理,以電子密度作為多電子體系量化處理的基本參量,使之包含原子與分子基態性質的所有信息[7].
幾乎在所有情況下,DFT的具體計算都涉及到Kohn-Sham軌道方程的求解[8]



從方程(1)中可以看出,Kohn-Sham方程引入了軌道概念.這樣,DFT方法與以波函數為基礎的常規從頭計算的方法存在同樣的缺陷,即用非完全基組對波函數的展開.對全基組(CBS)極限的研究將有利于了解DFT計算中給定泛函的系統偏差.Dunning等人[9-10]曾發現分子性質對基組選擇呈良好的單指數函數收斂行為.據此,人們便可估計出全基組極限及相應的計算性質[11-12]的系統偏差.
Geerlings等人[13]曾就電荷分布,化學活性指數,及相關性質的全基組收斂行為作了探討.本文運用7種常見交換相關泛函組合,以CO分子為例,對強成鍵體系CO分子的全基組收斂性質及不同泛函的表現進行了系統研究.
1 方法
DFT方法中一給定泛函的特定精確度可用其所求得一系列分子性質的全基組極限值來評估.本文所用性質為分子平衡鍵長re,解離能D0,和諧振基頻ωe.要求得全基組極限值,必須知道基組擴充的有效收斂規律.Dunning的擴展的調和相關價層雙ZETA((aug)-ccp-VDZ),三ZETA((aug)-ccp-VTZ),四ZETA((aug)-ccp-VQZ),和五ZETA((aug)-ccp-V5Z)基組即屬此類基組[14].對此類基組,人們發現[15]某種給定性質的計算值Q(X)能用如下單指數函數很好地表達:

其中,X為所用基組的ZETA數,Q(∞)為全基組極限值[9-10].一般說來,理論與試驗的誤差,ΔQapp,與基組大小的關系有兩種不同類型.第一種隨基組大小的增加而逐漸減小,故計算值隨基組的不斷增大而越來越靠近實驗值.第二種類型則有一極小點.在該極小點,計算值與實驗值非常接近,但仍與Q(∞)有很大差別.此類型常會被誤認為所用泛函的質量很好.
交換-相關泛函可表達為

其中,EX[ρ]與EC[ρ]分別是交換和相關能量密度泛函.如果它們僅是密度((r)本身的函數,則稱它們是局域的,否則是非局域的.本文將采用7種常用的交換-相關泛函.對交換泛函,我們采用Slater(S)[16],梯度修正的Becke 88(B)[17],及Hartee-Fock交換式[18],而對相關能泛函則采用Vosko-Wilk-Nusair(VWN)[19],梯度修正的Perdew 86(P86)[20],Perdew-Wang91(PW91)[21]及Lee-Yang-Parr(LYP)[22]公式.二者組合可得如下7種常見方法:SVWN,BLYP,B3LYP,BP86,B3P86,BPW91和B3PW91.
所有計算采用Gaussian′03 Window版[23]軟件在微機上完成.
2 結果與討論
2.1 平衡鍵長re
表1列出了用7種泛函組合與8種Dunning基組計算得到的平衡鍵長.實驗值為1.128 3 ?.由表中可見,計算值很好的收斂到CBS極值.然而,各個近似泛函的收斂行為卻不盡相同.所有梯度修正的泛函均表現為第一類收斂行為,而SVWN和以Hartee-Fock交換能為基礎的混合泛函方法,B3LYP,B3P86,和B3PW91則表現為第二類收斂行為.這表明用后面4種泛函,人們可以用較小基組得到較精確的平衡鍵長re值,但這并不說明這些計算方法較其他更好.一個例子是SVWN/cc-pVTZ,其re值為1.128 1 ?,與實驗值已非常接近,但該計算水平總的說來非常粗糙.
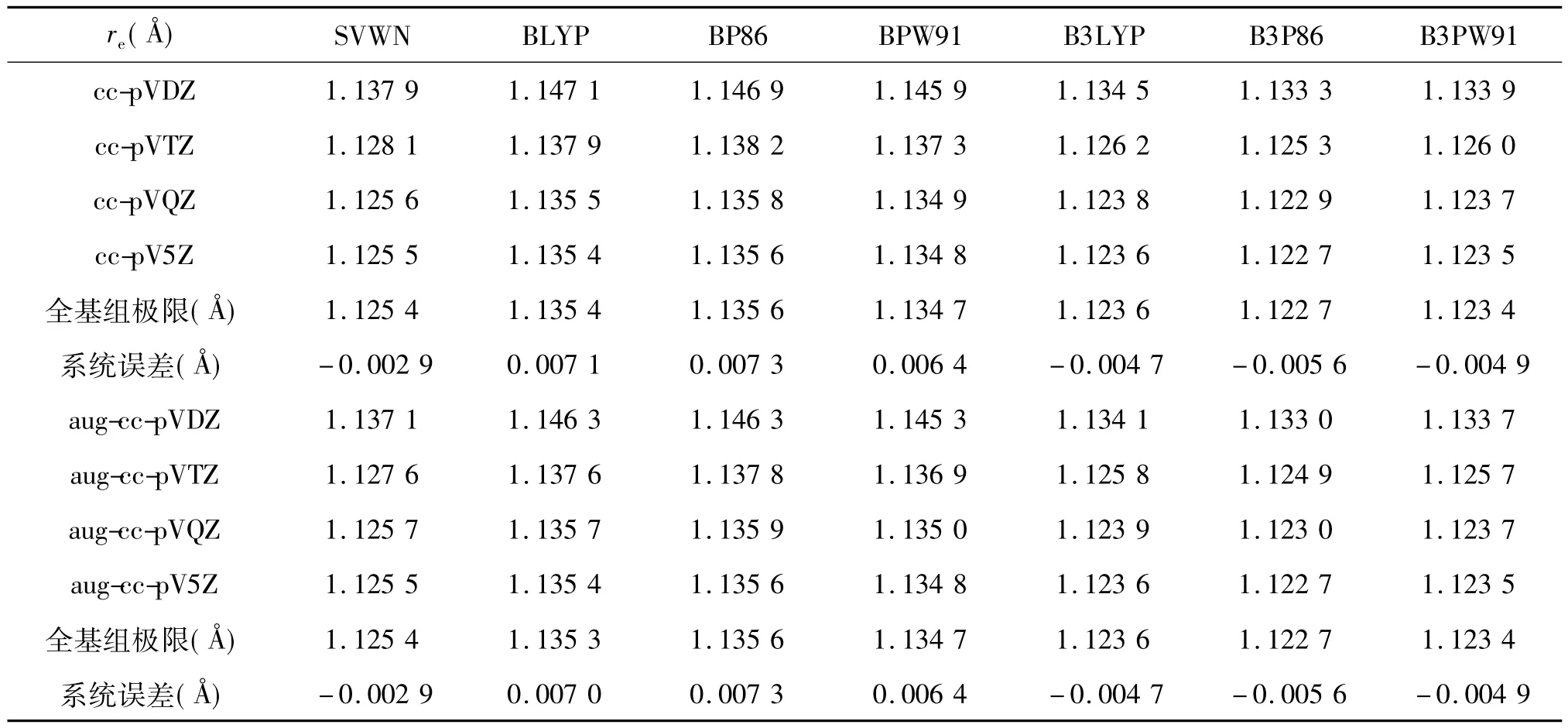
表1 用不同泛函和不同基組近似所得CO分子的平衡鍵長及各計算方法的全基組(CBS)極值和系統誤差單位:?
對給定基組下某性質計算值與全基組極限值的比較可用下式表達(ΔQbs/Q(∞))×100,其中ΔQbs為計算值與極限值之差.從表中可得,對(aug)-cc-pVDZ基組該值為±1%,(aug)-cc-pVTZ為±0.2%,(aug)-ccpVQZ為±0.02%,(aug)-cc-pV5Z為±0.005%.ZETA數越大,偏差率越小.另外,從表中可見,基組的擴展與否對平衡鍵長計算值的影響非常小,系統誤差的差別小于1 m?.與CBS極值相比,SVWN方法下的小擴展調和相關基組具有最大的偏差,但隨ZETA數增加而快速收斂至CBS極值.
從系統誤差來看,不同泛函組合的差別并不大,以SVWN的結果最小,低估2.9 m?.混合泛函平均低估5.0 m?,而梯度修正泛函平均高估7.0 m?.
2.2 諧振基頻ωeC
在表2中,我們列出了用上述7種方法、8種基組計算所得CO分子的諧振基頻,實驗值為217 0 cm-1[24].對于擴展的調和相關基組,計算值很好地收斂到CBS極值,其中梯度修正泛函表現為第一類收斂行為,而SVWN和混合泛函表現為第二類收斂行為.對非擴展的調和相關基組,由于從cc-pVQZ到cc-pV5Z有的沒有增加,有的反而降低,故用(4)式無法得到其CBS極值.
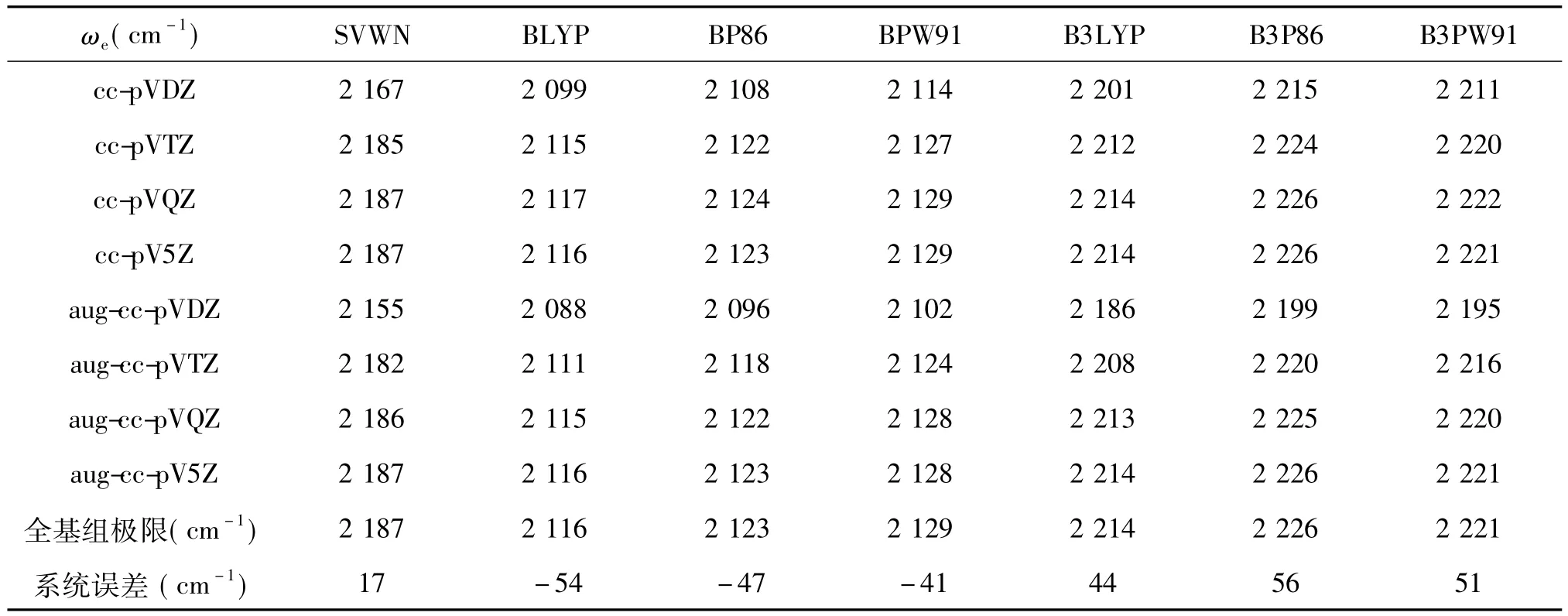
表2 用不同泛函和不同基組近似所得CO分子的諧振基頻ωe及各計算方法的全基組(CBS)極值和系統誤差單位:cm-1
如果考察ωe計算值的精度隨基組大小變化的關系,我們發現,跟平衡鍵長類似,與CBS極值相比較,SVWN泛函在較小基組時給出較大的偏差.隨基組ZETA數的增加,其值迅速收斂至CBS極值.
此外,同一基組不同泛函所得結果的差別并不大.對aug-cc-pVDZ基組,變化率為1.2%,aug-cc-pVTZ為0.2%,aug-cc-pVQZ為0.04%,aug-cc-pV5Z為0.008%.從系統誤差來看,以SVWN的結果最小,高估17 cm-1,混合泛函平均高估50 cm-1,而梯度修正泛函平均則平均低估50 cm-1.
2.3 離解能D0
表3報道了對CO分子離解能的計算值及其比較,其中實驗值為1 071.9 kJ/mol[24].我們觀察到了與諧振基頻一樣的收斂行為,即對擴展的調和相關基組,可以看到很好的收斂現象,而對另外的基組,由于從ccpVQZ到cc-pV5Z,計算值不增反降,用(4)式無法得到其CBS極值.對擴展的調和相關基組而言,混合泛函B3LYP及B3PW91表現為第一類收斂行為,其它則為第二類.各基組的偏差百分率分別為2.7%(aug-ccpVDZ),0.5%(aug-cc-pVTZ),0.06%(aug-cc-pVQZ),和0.01%(aug-cc-pV5Z).與平衡鍵長及諧振基頻一致,ZETA數越大,偏差值越小.從系統誤差而言,SVWN泛函的最大,高估186.6 kJ/mol,而梯度修正及混合泛函均表現出良好的精度.
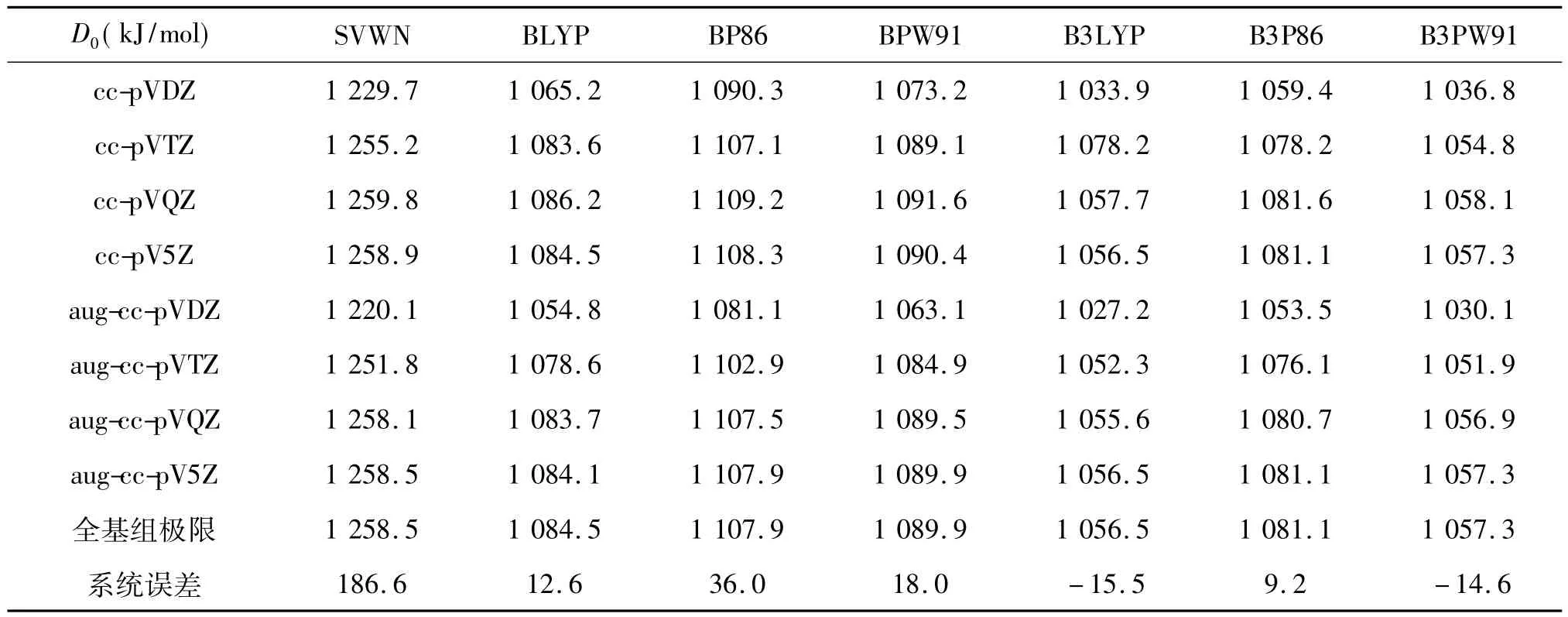
表3 用不同泛函和不同基組近似所得CO分子的離解能D0及各計算方法的全基組(CBS)極值和系統誤差單位:kJ/mol
2.4 討論
在現代密度泛函理論中,分子結構與性質的計算精度由兩個彼此獨立的因素決定,即交換-相關能的近似密度泛函形式及基組的相對大小(也就是由Kohn-Sham軌道組成的近似波函數的精確性).由于前者相對于后者更難精確地近似,DFT過去幾十年的發展均以前者為研究核心,代表性工作有LDA,GGA,meta-GGA,hybrid-GGA,double-hybrid GGA等.而對基組的作用特別是基組在計算結構和性質時表現出的收斂現象及其規律,仍然不見系統研究,而且在文獻中也沒有引起足夠的重視.本文以強成鍵體系CO分子為例,對3種常見物理化學性質,即平衡鍵長、諧振基頻和離解能,在計算時表現出的全基組收斂現象進行了系統研究,同時采用不同的泛函近似形式探討了不同的全基組收斂模式.發現不同的性質不同的泛函表現出不同的全基組收斂方式,反映了全基組收斂的復雜性.更有意思的是,從全基組收斂的角度來看,波函數計算的精確性與近似泛函的精確性并沒有相關性.這些結果表明,上述決定分子結構和性質計算精確度的兩個因素是彼此獨立的,沒有相關性.
3 結論
本文用密度泛函理論中7種常見泛函組合探討了用8種擴展和非擴展的調和相關基組來計算CO分子3種物理化學性質(即平衡鍵長,諧振基頻和離解能)時表現出的全基組收斂現象.并利用Dunning等人的單指數插補函數確定了這些性質的CBS極值.結果發現,不同的性質可能表現出截然不同的收斂行為.對平衡鍵長與諧振基頻而言,定域密度近似(LDA)和混合泛函方法表現為第二類收斂行為,而梯度修正則為第一類.對離解能,B3LYP和B3PW91兩個混合泛函方法表現出第一類,而其它則為第二類.令人吃驚的是,LDA方法給出了該分子平衡鍵長與諧振基頻的最好結果,而梯度修正(GGA)與混合泛函(hybrid GGA)方法的結果相當.對其它成鍵體系的研究正在進行中.
[1]PARR R G,YANG W.Density functional theory of atoms and molecules[M].New York:Oxford University Press,1989.
[2]DREIZLER R M,GROSS E K U.Density functional theory[M].New York:Springer-Verlag,Berlin Heidelberg,1990.
[3]POLITZER P,SEMINARIO J.Modern density functional theory;a tool for chemistry[M].Amsterdam:Elsevier,1995.
[4]PARR R G,YANG W.Density-functional theory of the electronic structure of molecules[J].Ann Rev Phys Chem,1995,46: 701-728.
[5]KOHN W,BECKE A D,PARR R G.Density functional theory of electronic structure[J].J Phys Chem,1996,100:12974-12980.
[6]LIU S B.Conceptual density functional theory and some recent developments[J].Acta Phys-Chim Sin,2009,25:590-600.
[7]HOHENBERG P,KOHN W.Inhomogeneous electron gas[J].Phys Rev B,1964,136:864-871.
[8]KOHN W,SHAM L J.Self-consistent equations including exchange and correlation effects[J].Phys Rev A,1965,140:1133-1138.
[9]WOON D E,DUNNING JR T H.Benchmark calculations with correlated molecular wave functions.Ⅰ.Multireference configuration interaction calculations for the second row diatomic hydrides[J].J Chem Phys,1993,99:1914-1929.
[10]PETERSON K A,KENDALL R A,DUNNING JR T H.Benchmark calculations with correlated molecular wave functions.Ⅱ. Configuration interaction calculations on first row diatomic hydrides[J].J Chem Phys,1993,99:1930-1944.
[11]黃雪,劉文龍,鐘愛國,等.雙核鎘配聚體及其衍生物熒光光譜發光機理的計算化學研究[J].湖南師范大學自然科學學報,2010,33(4):69-74.
[12]HUANG Y,ZHONG A G,LIU S B.Prediciting pKa values for singly and multiply substituted benzoic acids with density functional reactivity theory[J].湖南師范大學自然科學學報,2011,34(1):52-55.
[13]GEERLINGS P,DE PROFT F,LANGENAEKER W.Density functional theory:a source of chemical concepts and a costeffective methodology for their calculation[J].Adv Quant Chem,1999,33:303-328.
[14]DUNNING JR T H.Gaussian basis sets for use in correlated molecular calculations.Ⅰ.The atoms boron through neon and hydrogen[J].J Chem Phys,1989,90:1007-1023.
[15]MARTIN J M L.Energetics of stable molecules and reactive intermediates[M].NATO ASI Symposium Volume,M E Minas da Piedade and K K Irikura(Ed.),Dordrecht:Kluwer Academic Publishers,Dordrecht,1999.
[16]SLATER J C.A simplification of the hartree-Fock method[J].Phys Rev,1951,81:385-391.
[17]BECKE A D.Density-functional exchange-energy approximation with correct asymptotic behavior[J].Phys Rev A,1988,38: 3098-3100.
[18]BECKE A D.Density-functional thermochemistry.Ⅲ.The role of exact exchange[J].J Chem Phys,1993,98:5648-5652.
[19]VOSKO S H,WILK L,NUSAIR M.Accurate spin-dependent electron liquid correlation energies for local spin density calculations:a critical analysis[J].Can J Phys,1980,58:1200-1211.
[20]PERDEW J P.Density-functional approximation for the correlation energy of the inhomogeneous electron gas[J].Phys Rev B,1986,33:8822-8824.
[21]PERDEW J P.Electronic structure of solids[M].P Ziesche and H Eschrig(Ed.)Berlin:Akademie Verlag,1991.
[22]LEE C,YANG W,PARR R G.Development of the colle-salvetti correlation-energy formula into a functional of the electron density[J].Phys Rev B,1988,37:785-789.
[23]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 03,Revision E01,Gaussian,Inc.,Pittsburgh PA,2003.
[24]HUBER K P,HERZBERG G.Molecular spectra and molecular structureⅣ.Constants of diatomic molecules[M].Princeton:Van Nostrand,1979.
Basis Set Convergence Phenomenon in Density Functional Theory: CO as an Example of Strongly Bonded Systems
HUANG Ying*,LEI Zhi-dan,ZHANG Xiao-qing,LI Long
In this work we systematically investigate the basis set convergence phenomenon with seven commonly used approximate exchange-correlation energy density functionals and eight Dunning correlated basis sets for three physicochemical properties,equilibrium bond distance,first harmonic frequency,and dissociation energy,of the CO molecule.By employing Dunning′s single-exponent extrapolation approach,we have obtained the complete basis set limit for these properties.With the augmented correlation basis sets employed in this study,we found that the systematic error in the equilibrium bond distance is between 0.002 9(LDA)to 0.007 3 ?(BP86),that in the first harmonic frequency lies between 17(LDA)and 56(B3BP86)cm-1,and that in the dissociation energy falls between 12.6(BLYP)and 186.6 kJ/mol.These results shed lights on our systematic understanding of the basis set convergence phenomenon in the wave function.
density functional theory;basis set convergence;correlated basis set
O641.12+1
A
1000-2537(2011)05-0050-05
2011-05-20
湖南省自然科學基金資助項目(11JJ5065);湖南省大學生研究性學習和創新性實驗計劃項目(湘教通[2011]272號)
*通訊作者,E-mail:huanglingying@126.com
(編輯楊春明)
猜你喜歡
中學生數理化·高三版(2023年6期)2023-07-19 11:17:53
數學物理學報(2022年6期)2022-12-15 08:45:02
上海師范大學學報·自然科學版(2022年3期)2022-07-11 03:05:59
數學雜志(2021年6期)2021-11-24 11:12:00
中學生數理化(高中版.高二數學)(2021年5期)2021-07-21 02:14:46
數學年刊A輯(中文版)(2021年1期)2021-06-09 09:31:56
中等數學(2020年6期)2020-09-21 09:32:38
山東農業工程學院學報(2019年11期)2020-01-19 02:49:10
數學物理學報(2019年6期)2020-01-13 06:07:52
中等數學(2019年6期)2019-08-30 03:41:46
