光纖光度計測定鈾及鈾鈮合金中氮含量
2012-01-04 03:30:53武紅英李英秋
核化學與放射化學 2012年2期
關鍵詞:實驗
武紅英,李英秋,卞 敏
中國工程物理研究院,四川 綿陽 621900
鈾鈮合金的制備工藝中容易引入氮雜質,這種雜質由氣相表面層轉入凝聚相,以其單質(N2)及化合物(氮化鈾)形態存在,從而影響鈾鈮合金的物理和機械性能[1]。鑒于氮元素對鈾鈮合金特性的不利影響,建立鈾鈮合金中氮的測定方法很有必要。鈾鈮合金及鈾钚混合物中氮含量的測定一般有質譜法[2]、熱導法[3]、凱氏蒸餾分光光度法[2,4]。對于痕量或微量氮的分析大多采用凱氏蒸餾分光光度法。質譜法、熱導法對痕量或微量氮的測定精度低,回收率差。傳統的分光光度法測量模式是凱氏蒸餾分離出氨,對于低含量氮的測定,一般選擇百里酚作為顯色劑,氨溶液與酚形成穩定有色化合物[2,4,5-8],將待測化合物溶液放入樣品池,在一定波長下測其吸光度,從而計算出氮的含量。這樣對于強放射性樣品的分析,操作相當煩瑣,不便于儀器維護。近些年來,光纖技術的引入使得紫外光譜儀器有了迅速的發展,待測物脫離了樣品池的限制,采樣方式變得更為靈活,如利用光纖探頭掃描樣品溶液就可以遠離樣品把信號傳到光譜儀器,操作便捷、維護方便,特別適合強放射性樣品的測量及操作[9-11]。本工作擬在前人研究基礎上,采用光纖光度計對傳統鈾鈮合金中氮含量的測量模式進行改進,并與傳統分光光度法作比較。
1 實驗部分
1.1 試劑與材料
本實驗中所用水均為蒸餾水。鹽酸,超純,上海來澤精細化學品廠;氫氧化鈉溶液,分析純,成都化學試劑廠;氯化銨,高純,上海試劑一廠;磷酸氫二鈉、丙酮,分析純,均為成都市聯合化工試劑研究所;麝香草酚,分析純,重慶東方試劑廠;次氯酸鈉儲備溶液(w(Cl)=0.1%,0.3%),自制;顯色劑(1+5):百里酚溶液+磷酸氫二鈉溶液。
氮標準儲備溶液:稱取0.381 9 g優級氯化氨(預先在105 ℃干燥1 h),置于100 mL燒杯中,加水溶解,移入1 000 mL容量瓶中,用水稀釋至刻度,混勻,此溶液1 mL含100 μg氮。
氮標準溶液:移取10.00 mL氮標準儲備溶液,置于1 000 mL容量瓶中,用水稀釋至刻度,混勻。此溶液1 mL含1 μg氮。
氬氣,純度99.99%。氬氣先經過一個裝有氫氧化鈉溶液(w=10%)的洗氣瓶,再經過水洗、濃硫酸脫水、洗氣瓶洗滌后使用。
純鈾及鈾鈮合金基體溶液(20 g/L):稱取1 g純鈾及鈾鈮合金試樣置于100 mL燒杯中,加入50 mL鹽酸(V(HCl)∶V(H2O)=1∶1)在電熱板上慢慢溶解,試樣溶解后再加入2 mL雙氧水,待雙氧水揮發完全,用鹽酸稀釋至50 mL刻度。
鈾鈮合金切屑試樣,由生產工藝提供。
1.2 儀器
Cary50光纖光度計,美國瓦里安公司;玻璃蒸餾儀,自制。AE100電子天平,感量為0.01 mg,瑞士梅特勒公司。
1.3 實驗方法
稱取(20±2)mg鈾鈮合金切屑試樣于20 mL燒杯中,加入1 mL的鹽酸(V(HCl)∶V(H2O)=1∶1)及兩滴雙氧水在電熱板上蒸至近干,讓過量的雙氧水揮發完全,轉入蒸餾儀中,加入1 mL氫氧化鈉溶液(w=50%)和適量水,通氬氣進行蒸餾。當餾出液達(7.0±0.5)mL時,停止蒸餾,加入2滴(0.04 mL)次氯酸鈉溶液于10 mL吸收容量瓶,充分搖勻后放置5 min,加入2.0 mL顯色劑,用水稀釋至刻度,搖勻后放置40 min于660 nm波長處,以水或試劑空白為參比,用20 mm光纖探頭分別測定其吸光度。從工作曲線上直接換算出氮的含量。
2 結果與討論
2.1 吸收光譜
移取一定量氮標準溶液于10 mL吸收容量瓶,按照實驗方法加入次氯酸鈉溶液及百里酚顯色劑。氨與次氯酸鈉生成一氯氨,一氯氨與百里酚于強堿溶液中生成吲哚百里酚[12-14]:
NH3+NaOCl= NH2Cl+NaOH
NH2Cl+C10H13OH+2NaOCl=
C10H12ONCl+2NaCl+2H2O
C10H12ONCl+C10H13OH=C20H24ONOH+HCl
生成的吲哚百里酚溶液于Cary50光纖光度計上進行掃描,其吸收光譜示于圖1。由圖1可知,氨與酚生成的化合物在波長660~670 nm處吸光度最高,文獻[8,13]報道在625~640 nm、665~670 nm處吸光度最高,該法與傳統分光光度法峰形基本一致,能更便捷掃描吸收光譜,實驗確定測定波長為660 nm。
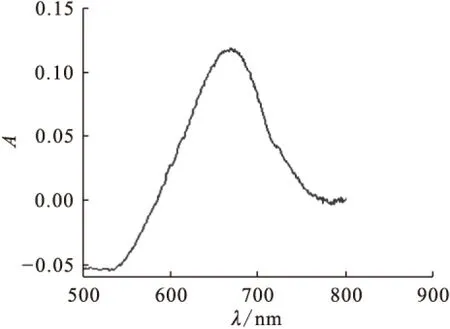
圖1 吲哚百里酚的吸收光譜
2.2 條件實驗
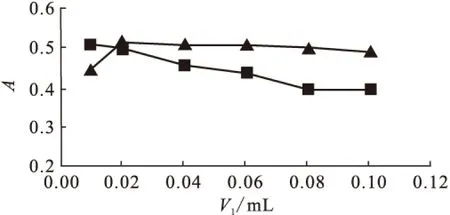
圖2 次氯酸鈉用量實驗
2.2.1次氯酸鈉用量對結果的影響 用w(Cl)=0.3%和0.1%的2種次氯酸鈉溶液分別作試劑用量(V1)實驗,實驗結果示于圖2。由圖2看出:采用w(Cl)=0.3%的次氯酸鈉溶液時,隨著其用量增加,吸光度下降很快;而采用w(Cl)=0.1%次氯酸鈉溶液,則隨著其用量增加,吸光度降低較慢,與文獻[7]報道一致。實驗采用w(Cl)=0.1%的次氯酸鈉溶液0.04 mL(2滴)。

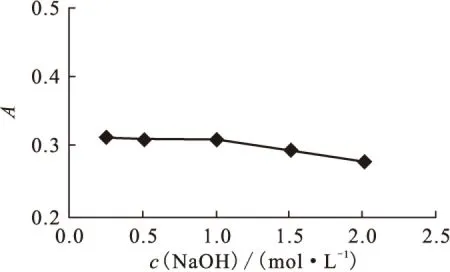
圖3 氫氧化鈉濃度對吸光度測量的影響
2.2.3有效氯含量對結果的影響 采用不同濃度的次氯酸鈉溶液作實驗,測量結果示于圖4。由圖4可以看出,次氯酸鈉溶液有效氯濃度在0.1%~0.15%范圍內波動不大。實驗確定次氯酸鈉溶液有效氯濃度w(Cl)=0.1%~0.14%。
2.2.4氯化時間和溫度對結果的影響 在不同溫度下加入次氯酸鈉溶液后考察放置時間(t1)對結果的影響,結果示于圖5。由圖5可以看出:當室溫17 ℃時,放置10 min后吸光度才穩定;而當室溫25 ℃時,放置5 min后吸光度就穩定了。實驗確定放置時間為10 min。
2.2.5顯色劑用量對結果的影響 考察顯色劑用量(V2)對結果的影響,結果示于圖6。由圖6可知,實驗選擇顯色劑用量V2=2 mL為宜,這與文獻[4]報道一致。
2.2.6顯色時間對結果的影響 分別在17 ℃和25 ℃時考察顯色時間(t2)對結果的影響,結果示于圖7。由圖7可以看出,溫度影響顯色反應的速度。因此確定25 ℃下放置20 min。如果實驗室溫度低于25 ℃則放置40 min為宜,這與文獻[4]報道一致。
2.3 標準曲線
分別移取0.00、0.40、0.80、1.20、1.60、2.00 mL氮標準溶液于20 mL燒杯中,各加入2滴鹽酸溶液分別轉入蒸餾儀中,按1.3節方法分析。以含氮量為橫坐標、吸光度為縱坐標繪制不含合金基體的標準曲線,示于圖8。
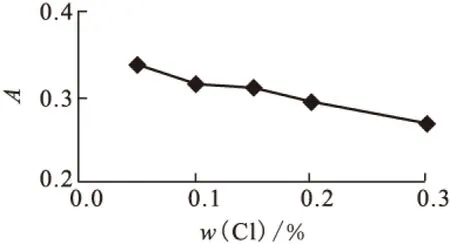
圖4 有效氯含量對結果的影響
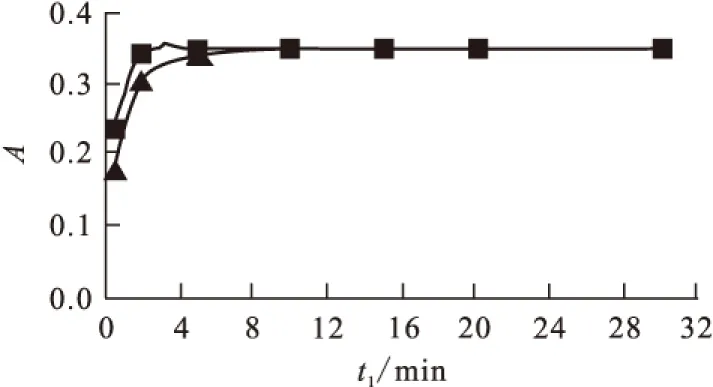
圖5 氯化時間和溫度對結果的影響
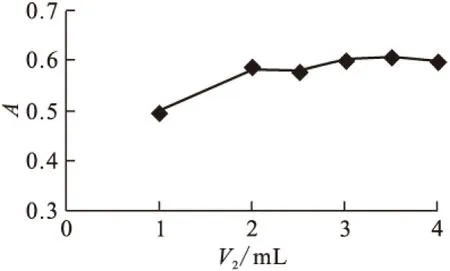
圖6 顯色劑用量對吸光度的影響
分別移取1 mL鈾鈮合金基體溶液于6個20 mL燒杯中,在電熱板上加熱至近干,冷卻至室溫,再分別移取0.00、0.40、0.80、1.20、1.60、2.00 mL氮標準溶液于上述燒杯中,分別轉入蒸餾儀中,按1.3節方法分析,扣除鈾鈮合金基體溶液吸光度并繪制含合金基體的標準曲線,示于圖8。從圖8中可以看出,這兩條標準曲線基本一致,實際生產分析中可用不含合金基體的標準曲線代替后者。
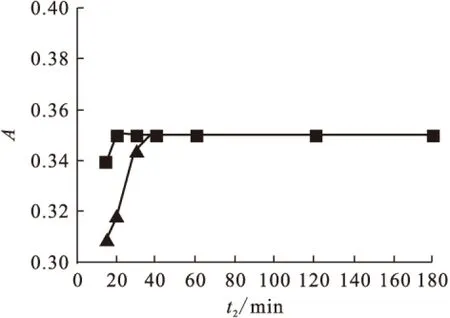
圖7 顯色時間對吸光度的影響
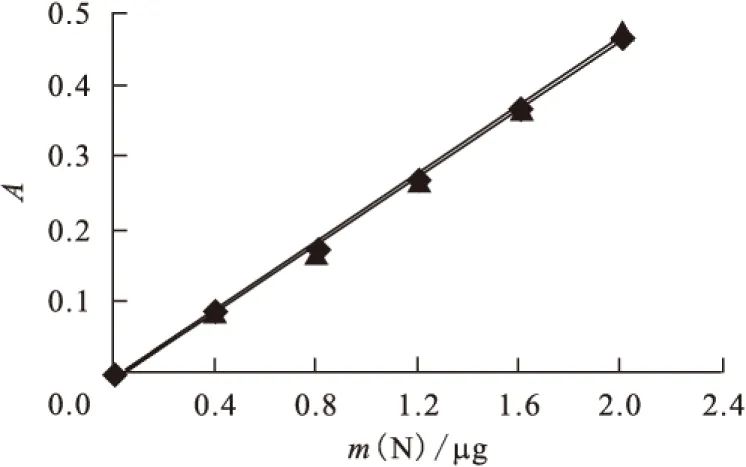
圖8 標準曲線
2.4 方法的檢測下限

2.5 回收實驗
分別移取1 mL鈾鈮合金基體溶液于20 mL燒杯中,并分別加入0.8、1.2、1.6 μg氮標準溶液作回收實驗,結果列入表1。由表1可知:加入0.8 μg氮時,回收率為98%~101%;加入1.2 μg氮時,回收率為88%~100%;加入1.6 μg氮時,回收率為100%~106%。
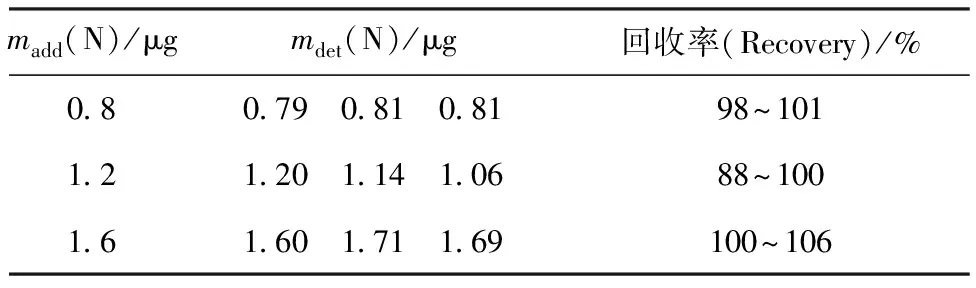
表1 回收實驗測定結果
2.6 精密度實驗
分別對不同氮含量的鈾鈮合金切屑試樣作精密度實驗,結果列入表2。由表2可知,對于含氮量為28 μg/g的鈾鈮合金切屑試樣,測量的相對標準偏差小于15%(n=6)。
2.7 與傳統分光光度法、熱導法結果比對
分別對鈾鈮合金切屑試樣,采用光纖光度法與傳統分光光度法、熱導法作了比對,結果列入表3。由表3可知,3種方法測量結果基本一致,對于含氮量大于30 μg/g以上的鈾鈮合金切屑試樣,熱導法測量結果略高。這是由于化學法在溶解蒸餾過程中并不能使樣品中的氮完全釋放出來[2-3],在工藝允許的誤差范圍內,光纖光度法可以滿足實際生產要求。

表2 精密度實驗測定結果
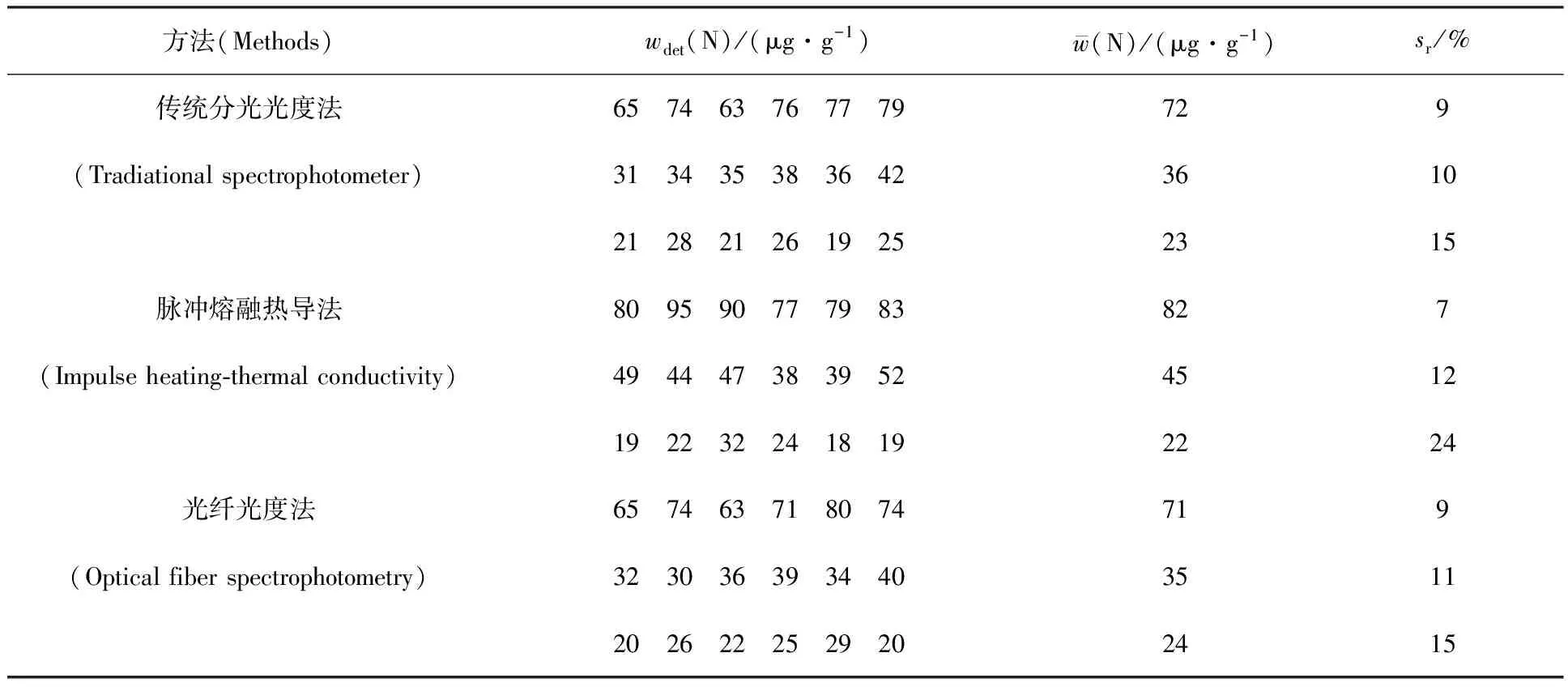
表3 鈾鈮合金試樣中氮的測量結果比對
3 結 論
采用光纖光度計研究了百里酚顯色測定鈾及鈾鈮合金材料中氮的影響因素條件實驗。結果表明,在w(Cl)=0.1%~0.14%的溶液中,氨與百里酚形成藍色高靈敏度化合物,該化合物的最大吸收波長為660 nm,在常溫下放置40 min進行測量,結果穩定,方法檢測限為20 μg/g,測量相對標準偏差小于15%,方法回收率為88%~106%。與傳統分光光度法實驗條件結果對照,實驗條件一致。并與傳統分光光度法、熱導法進行了比對,測量結果基本一致,該法在測量模式上較之傳統的分光光度法具有更簡便、快速、準確的特點。
[1]伯克J J.鈾合金物理冶金[M].石琪,譯.北京:原子能出版社,1983:67.
[2]Sinclair V M,Davies W,Melhuish K R.Determination of Nitrogen in Uranium-Plutonium Dioxides[J].Talanta,1965,12(9): 841-845.
[3]吳倫強,鄒樂西,向方壽,等.同時測定鈾鈮合金中微量氧和氮[J].理化檢驗-化學分冊,2001(3):8-12.
[4]Rizvi G H,Natrajan P R.Determination of Nitrogen in UO2by Kjeldahl Spectrophotometry[J].J Radioanal Nucl Chem,1986,102(2): 515-523.
[5]洪鯤.百里酚光度法測定水中硝酸鹽氮[J].環境研究與監測,2004,17(3):18-28.
[6]黃根華,施小春.鈦及鈦合金中氮的測定[J].星河科技,1992(3):14-18.
[7]Weatherburn M W.Phenol-Hypochlorite Reaction for Determination of Ammonia[J].Anal Chem,1967,39(8): 971-974.
[8]Solorzano L.Determination of Ammonia in Natural Waters by the Phenol Hypochlorite Methods[J].Limnol Oceanogr,1969,14: 799-801.
[9]Danigel H.光纖測量技術[J].高蔚,譯.現代計量測試,1996(3):58-61.
[10]莊維新,田果成,葉國安,等.光纖光導分光光度計:CNIC-1280[R].北京:中國原子能科學研究院,1998.
[11]游文海.光纖傳感雙波長導數光譜測定鈥、鉺[J].稀土,1995,16(3):31-35.
[12]郁沁明.堿分離-百里酚顯色光度法于鋼鐵中化合氮分析之研究[J].分析實驗室,2007,26(12):62-65.
[13]Hata N,Teraguchi K,Yamaguchi M,et al.Spectrophotometric Determination of Ammonim-Nitrogen After Preconcentration as Indothymol on a Glass-Fiber Filter in the Presence of a Cationic Surfactant[J].Microchim Acta,1992,106: 101-108.
[14]Duke S,Cullaj A.An Optical Procedure for Ammoniacal Nitrogen Analysis in Natural Waters Using Indophenol Blue Methods[J].Natura Montenegrina,Podgorica,2010,9(3): 743-751.
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
