表達紅色熒光蛋白重組柯薩奇病毒B組3型基因組穩定性分析
2012-01-23 01:12:38趙文然鐘學寬張淑娟沃曉嫚張中海王博鐘照華
微生物與感染 2012年1期
趙文然,鐘學寬,張淑娟,沃曉嫚,張中海,王博,鐘照華
1. 哈爾濱醫科大學細胞生物學教研室,哈爾濱 150086; 2. 哈爾濱醫科大學地方病防治研究中心,哈爾濱 150086; 3. 哈爾濱醫科大學微生物學教研室,哈爾濱 150086
柯薩奇病毒B組(coxsackie virus B,CVB)是引起病毒性心肌炎的主要病原體,在一部分患者CVB感染可發展為擴張型心肌病[1-3]。在研究CVB感染時,通常要對細胞或實驗動物的病毒感染情況進行定性或定量分析,或需要明確病毒在體內復制的部位及強度。對病毒感染的定性分析方法通常為觀察細胞病變(cytopathic effect, CPE),而定量分析常用方法包括測定半數組織培養感染量(50% tissue culture infective dose, TCID50)、噬斑形成單位(plaque forming unit, PFU),或通過反轉錄-聚合酶鏈反應(reverse transcriptase-polymerase chain reaction, RT-PCR)測定感染細胞內的病毒核酸含量[4-7]。以上幾種方法雖然很可靠,但除了對病毒感染細胞進行形態觀察以外,其他方法均很耗時,且操作復雜。近年來,廣泛應用的生物標記技術既可對病毒感染進行定性、定量分析,又可對病毒在體內感染過程進行示蹤[8-11]。熒光報告基因標記是目前常用的生物標記方法之一,其中編碼紅色熒光蛋白(red fluorescent protein,RFP)的報告基因mCherry可用于研究多種病原體感染。mCherry的波長相對較長,可避免體內自發綠熒光的干擾[10,11]。對RNA病毒而言,由于RNA聚合酶缺乏糾錯功能,病毒在復制過程中易產生基因突變[12-14]。這一特點可能影響重組RNA病毒基因組的穩定性[15]。因此,如果要通過紅色熒光蛋白的表達對柯薩奇病毒B組3型(coxsackie virus B3,CVB3)的復制進行定性和定量分析,則有必要了解含mCherry報告基因的重組CVB3基因組的穩定性。
1 材料和方法
1.1 材料
1.1.1 質粒、細胞及主要試劑 pMKS1由J. Lindsay Whitton教授(The Scripps Research Institute,California)惠贈。質粒peGFP-N1購自Clontech公司。重組質粒pCVB3-mCherry(圖1)攜帶嗜心肌的CVB3 H3毒株基因組全長cDNA及紅色熒光蛋白mCherry基因編碼序列由哈爾濱醫科大學微生物學教研室構建。該重組質粒的構建方法見文獻[14],即以pMKS1為基礎,將序列兩端含內切酶SfiI識別序列的mCherry基因插入CVB3基因組5′端。mCherry的激發波長為580 nm,發射波長為610 nm。HeLa細胞由哈爾濱醫科大學微生物學教研室保存。質粒小量提取試劑盒、DNA小量膠回收試劑盒購自Axygen公司。DL2000 DNA Ladder、DL15000 DNA Ladder、pMD19-T Simple Vector、LATaq聚合酶和dNTP購自TaKaRa公司。真核轉染試劑Lipofectamine 2000購自Invitrogen公司。胎牛血清(fetal calf serum,FCS)購自以色列Biological Industries公司(Cat: 04-001-1A;Lot: 215356)。中性紅細胞染色液購自Sigma公司。細胞核染料Hoechst 33342購自Invitrogen公司,激發波長為350 nm,發射波長為480 nm。

圖1 CVB3-mCherry的結構Fig.1 The structure of CVB3-mCherry
1.1.2 主要儀器 主要儀器包括Mastercycler gradient PCR儀(Eppendorf)和Axiovert 200相差倒置熒光顯微鏡(Carl Zeiss)。
1.1.3 主要軟件及網站 Gene runner用于引物設計及多重序列比對;數據庫http://www.ncbi.nlm.nih.gov/BLAST用于基因組結構查詢及序列比對。
1.2 方法
1.2.1 重組質粒pCVB3-mCherry的擴增與純化 取保存于-80 ℃含重組質粒pCVB3-mCherry的菌液適量,接種于5 ml含氨芐西林的LB培養基中,于37 ℃搖床振搖12~16 h(180 r/min),可見液體渾濁。取1.5 ml菌液加入離心管中,122 000g離心1 min,棄上清液。用質粒小量提取試劑盒提取質粒,檢測濃度及純度,-20 ℃保存備用。
1.2.2 pCVB3-mCherry在HeLa細胞中的瞬時表達 于24孔細胞培養板中培養HeLa細胞,24 h后選取生長狀態良好且生長至70%~90%單層細胞,用OPTI-MEMI培養基稀釋脂質體和質粒pCVB3-mCherry。轉染細胞時,脂質體與質粒的比為2 μl∶0.8 μg;轉染時設陽性對照(peGFP-N1轉染細胞)、陰性對照及未經任何處理的HeLa細胞對照;置培養板于37 ℃、 5% CO2培養箱中培養,24 h后觀察熒光表達及細胞病變情況。
1.2.3 病毒的收獲及傳代 用pCVB3-mCherry轉染HeLa細胞,當70%~90%細胞出現病變時,收獲細胞,反復凍融3次,于2 000g、4 ℃離心10 min,收集上清液,稱為第1代病毒,儲存于-80 ℃。用第1代病毒感染HeLa細胞,待70%~90%細胞出現病變時,收獲細胞,反復凍融3次,離心后收集上清液,稱為第2代病毒。以此類推,將病毒連續傳至第6代。
1.2.4 噬斑形成實驗及病毒毒力測定 將HeLa細胞以2.5×105個/孔接種于6孔細胞培養板中,于37 ℃、5% CO2培養箱中培養18~24 h。當細胞長至單層無縫隙時,用DMEM倍比稀釋 (10倍稀釋) 的病毒接種。每孔加入病毒稀釋液650 μl,置37 ℃、5% CO2培養箱中培養1 h,棄病毒稀釋液;每孔加入2 ml固體培養基(含5% FCS的2×DMEM和1.6% 瓊脂糖,1∶1配制),放入濕盒,置37 ℃、5% CO2培養箱,30 min后倒置培養72 h。鏡下觀察熒光表達及CPE,根據CPE情況進行染色。于細胞培養板中加入0.05%中性紅染色液,室溫作用1 h,棄染液。挑選培養板中噬斑未融合且噬斑個數適中的培養孔,鏡下計數噬斑個數,按以下公式計算病毒毒力:PFU/ml=每個稀釋濃度噬斑的平均值/(病毒的稀釋濃度×每孔病毒接種量)。
1.2.5 重組毒株的純化及穩定性評價 取第1代病毒用于噬斑形成實驗。挑取單個噬斑,接種于HeLa細胞進行擴增(100 ml規格培養瓶),于37 ℃、5% CO2培養箱中靜置培養;當90%細胞出現CPE時,將細胞反復凍融3次,收獲病毒,此為純化后的CVB3-mCherry第1代病毒。以同樣方法純化第2~6代病毒。分別提取每代病毒總RNA,用RT-PCR擴增重組毒株中的mCherry報告基因及CVB3基因組部分序列,經0.7%瓊脂糖電泳觀察擴增產物片段大小。
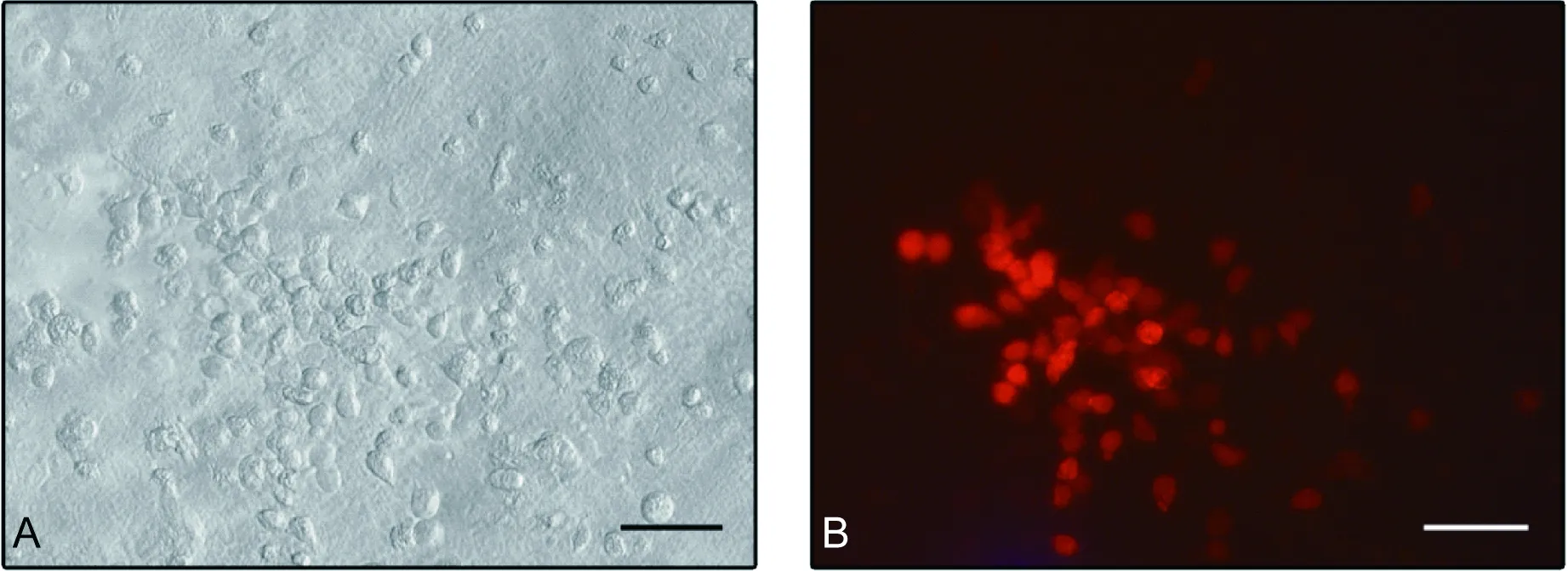
HeLa cells transfected with pCVB3-mCherry in light microscopy (A) and fluorescence microscopy (B). Scale bar, 100 μm.
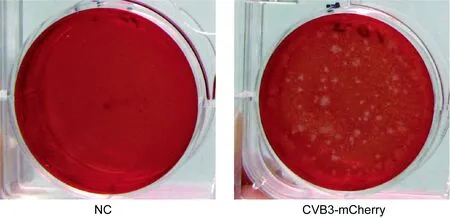
NC:HeLa cells without CVB3-mCherry infection; CVB3-mCherry: HeLa cells infected with the first passage of CVB3-mCherry.

A: RT-PCR strategy to analyze the stability of CVB3-mCherry. B: RT-PCR results. C : Cytopathic effect of CVB3-mCherry in HeLa cells in light and fluorescence microscopy. Panel 0 represents HeLa cells without virus infection. Panel 2-6 represent HeLa cells infected with the 2-6 passages of CVB3-mCherry.
PCR引物序列如下:上游引物(P1)5′-GGCGGCAGTGTGTCGTAACGGGC- AAC-3′(CVB3的5′ UTR區),下游引物(P2)5′-GCGTGGTTCTGTGAACTTGCCCGGG-3′(CVB3的VP4區)。如果重組病毒基因組攜帶mCherry基因,擴增片段預期為1 160 bp;如果重組病毒基因組丟失報告基因mCherry,擴增片段應為455 bp。用pCVB3-mCherry、pMKS-1作為擴增模板的陽性對照,ddH2O為陰性對照。將擴增產物各片段用凝膠回收法純化,進行TA克隆,測序(北京英駿公司)后分析目的片段丟失規律。測序引物為PMD19T載體通用引物,引物序列為M13F-47(CGCCAGG-GTTTTCCCAGTCACGAC)和M13R-48(AGCG-GATAACAATTTCACACAGGA)。
2 結果
2.1 pCVB3-mCherry在HeLa細胞中的瞬時表達
用pCVB3-mCherry轉染HeLa細胞,培養60 h 后,光學顯微鏡下可見CPE,熒光顯微鏡下可見細胞內紅色熒光(圖2),表明CVB3-mCherry可感染細胞并在細胞內復制,可用于CVB感染的體外研究。
2.2 CVB3-mCherry的毒力
用重組質粒pCVB3-mCherry轉染HeLa細胞,轉染60 h后檢測到紅色熒光,收獲第1代重組病毒,用噬斑形成實驗測得病毒毒力為9.3×106PFU/ml(圖3)。
2.3 CVB3-mCherry的穩定性
提取各代CVB3-mCherry基因組RNA,用于RT-PCR擴增。結果(圖4B)顯示,第2代和第3代重組病毒的擴增產物于1 160 bp處有較亮的條帶,455 bp處未見到條帶,但可見長度<455 bp的片段。隨著傳代次數遞增,擴增產物中的1 160 bp片段顯示的條帶逐漸模糊;同時,長度<455 bp的未知片段則隨病毒傳代次數的增加而變得清晰。此結果表明,報告基因mCherry的丟失開始于重組病毒的第2代;在mCherry基因片段丟失的同時,部分CVB3基因片段也隨之丟失。然而,第2~6代重組病毒所致的CPE沒有顯著區別(圖4C)。
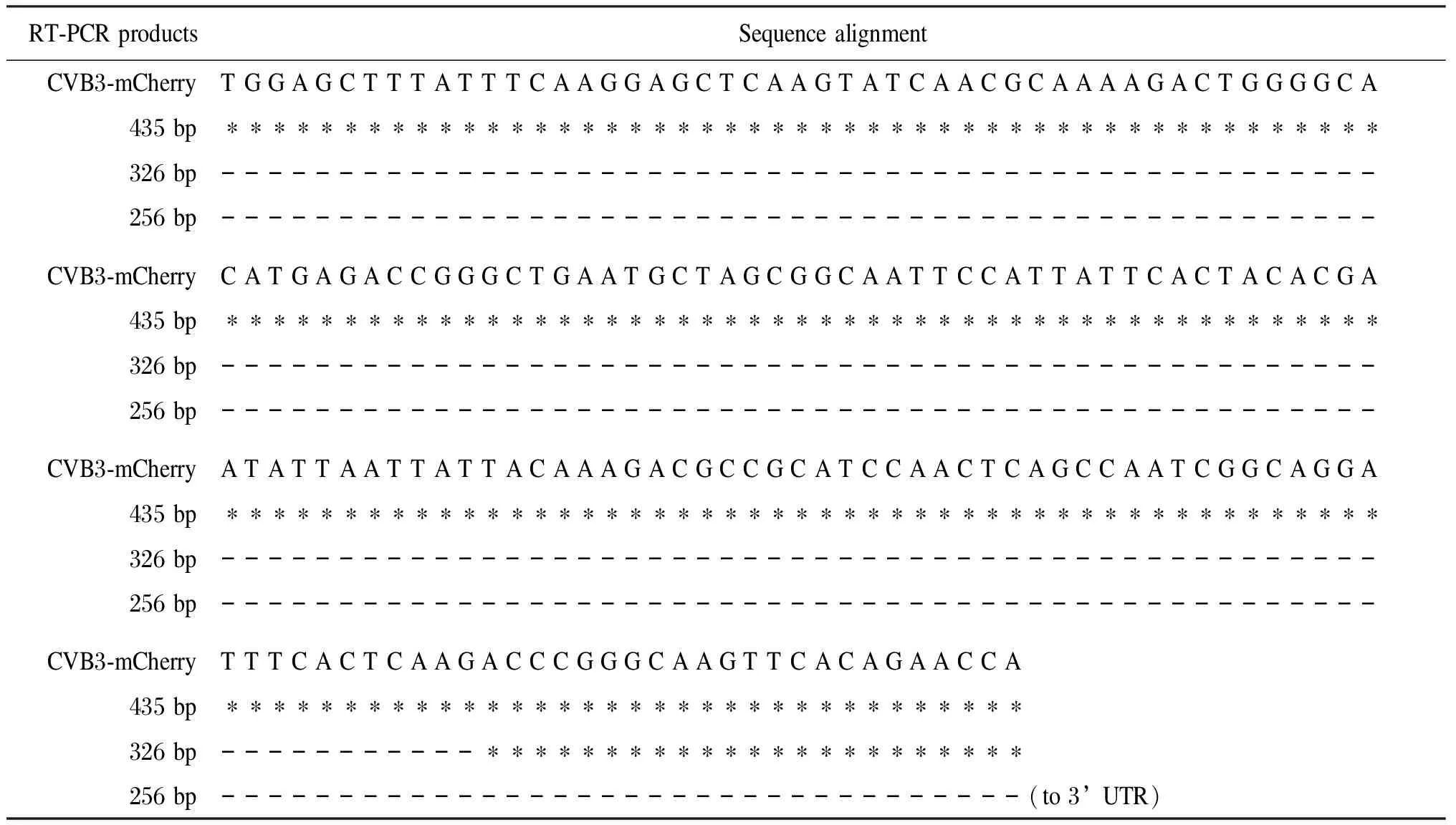
將RT-PCR擴增產物中3個長度<455 bp的小片段回收純化,經TA克隆后測序,與重組質粒pCVB3-mCherry進行序列比對。其中RT-PCR獲得的3個小片段長度分別為435 bp、326 bp和256 bp。435 bp片段的產生是由于重組病毒丟失了包括插入的SfiI酶切位點及mCherry全序列,并因此導致病毒開放讀碼框架(open reading frame,ORF)移位,但無病毒基因序列丟失;326 bp片段的產生是由于丟失了mCherry及病毒的VP4區部分序列;而256 bp片段的產生則是mCherry和CVB3 ORF全部丟失的結果(表1、圖5)。
3 討論
CVB為單股正鏈RNA病毒,能引起人類多種疾病,可侵犯心臟、脊髓、胰腺、腎臟、腦等多個重要器官,是病毒性心肌炎的主要病原體[1-3]。目前,對CVB感染進行定性和定量的診斷方法仍以形態學觀察、測定PFU及RT-PCR為主。近年來熒光報告基因標記也廣泛用于病毒檢測。該方法更便捷,可用于病毒感染的定量分析及動態觀察,但目前對于攜帶熒光報告基因的重組CVB基因組的穩定性仍缺乏了解。
本研究結果表明,重組體CVB3-mCherry能在HeLa細胞內復制并產生明顯的CPE,可通過觀察紅色熒光蛋白表達情況來評價CVB的感染情況。此方法快速、簡便。與其他普通的紅色熒光蛋白相比,mCherry的優點在于有較長的發射波長、單體形式、在細胞內熒光轉換效率高,因而具有較高的信噪比,更易被檢測到;其次,檢測時無需底物,只需激發光,既方便又節約成本[10,11]。

Red thin lines represent the locations of the fragments obtained from the 2-6 passages of CVB3-mCherry.

表1 重組病毒CVB3-mCherry與其突變體基因序列分析結果比對Fig.1 Sequence alignment between the recombinant CVB3-mCherry and its mutated progenies

(續表)
(續表)
* represents the nucleotide that is retained in CVB3-mCherry; - represents the nucleotide that has been lost from CVB3-mCherry.
基因組不穩定是小RNA病毒的共同特征[12-14]。有研究報道,與報告基因增強型綠色熒光蛋白(enhanced green fluorescent protein, eGFP)重組后,CVB基因組變得不穩定,在傳代過程中會丟失報告基因及部分CVB基因序列[15]。因此推測,隨著傳代次數的增加,CVB3-mCherry毒株的穩定性也可能有所變化。本研究表明,從第2代開始,重組病毒中就發生了報告基因丟失。丟失的序列包括插入的報告基因mCherry、CVB的5′ UTR區及部分VP4區。而第4~6代的重組病毒更不穩定,其中未發生基因片段丟失的病毒數量逐漸減少,表現為PCR產物中1 160 bp處的條帶越來越模糊,而發生基因丟失的病毒數量逐漸增加。序列分析表明,由于核酸序列丟失導致病毒ORF移位,因此會產生致死性突變株,使病毒失去原有的感染性。
本研究結果表明,與含報告基因eGFP的重組CVB3類似[15],重組CVB3-mCherry基因組也具有不穩定的特點。雖然從第2~6代重組病毒所致的CPE看不出顯著變化,但包括CVB3自身基因片段在內的基因序列丟失從第2代就已存在。因此,應用時病毒傳代次數不宜超過2代;長期應用則需重新用重組質粒轉染敏感細胞以收獲病毒。此外,應通過噬斑形成實驗純化CVB3-mCherry毒株并測定其毒力。盡管如此,該重組體仍不失為研究CVB的方便且可靠的工具。
[1] Pinkert S, Westermann D, Wang X, Klingel K, D?rner A, Savvatis K, Gr?ssl T, Krohn S, Tsch?pe C, Zeichhardt H, Kotsch K, Weitmann K, Hoffmann W, Schultheiss HP, Spiller OB, Poller W, Fechner H. Prevention of cardiac dysfunction in acute coxsackievirus B3 cardiomyopathy by inducible expression of a soluble coxsackievirus-adenovirus receptor [J]. Circulation, 2009, 120(23): 2358-2366.
[2] Rutschow S, Leschka S, Westermann D, Puhl K, Weitz A, Ladyszenskij L, Jaeger S, Zeichhardt H, Noutsias M, Schultheiss HP, Tschope C, Pauschinger M. Left ventricular enlargement in coxsackievirus-B3 induced chronic myocarditis-ongoing inflammation and an imbalance of the matrix degrading system [J]. Eur J Pharmacol, 2010, 630(1-3): 145-151.
[3] Luo H, Wong J, Wong B. Protein degradation systems in viral myocarditis leading to dilated cardiomyopathy [J]. Cardiovasc Res, 2010, 85(2): 347-356.
[4] Mitsuya H, Broder S. Inhibition of the in vitro infectivity and cytopathic effect of human T-lymphotrophic virus type III/lymphadenopathy-associated virus (HTLV-III/LAV) by 2′,3′-dideoxynucleosides [J]. Proc Natl Acad Sci USA, 1986, 83(6): 1911-1915.
[5] Van Houten N, Bouchard PE, Moraska A, Huber SA. Selection of an attenuated Coxsackievirus B3 variant, using a monoclonal antibody reactive to myocyte antigen [J]. J Virol, 1991, 65(3): 1286-1290.
[6] Lyden DC, Olszewski J, Feran M, Job LP, Huber SA. Coxsackievirus B-3-induced myocarditis. Effect of sex steroids on viremia and infectivity of cardiocytes [J]. Am J Pathol, 1987, 126(3): 432-438.
[7] Piqueur MA, Verstrepen WA, Bruynseels P, Mertens AH. Improvement of a real-time RT-PCR assay for the detection of enterovirus RNA [J]. Virol J, 2009, 6: 95.
[8] Luker GD, Luker KE. Optical imaging: current applications and future directions [J]. J Nucl Med, 2008, 49(1): 1-4.
[9] Wong J, Zhang J, Si X, Gao G, Mao I, McManus BM, Luo H. Autophagosome supports coxsackievirus B3 replication in host cells [J]. J Virol, 2008, 82(18): 9143-9153.
[10] Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins [J]. Nat Methods, 2005, 2(12): 905-909.
[11] Campbell EM, Perez O, Melar M, Hope TJ. Labeling HIV-1 virions with two fluorescent proteins allows identification of virions that have productively entered the target cell [J]. Virology, 2007, 360 (2): 286-293.
[12] Drake JW, Holland JJ. Mutation rates among RNA viruses [J]. Proc Natl Acad Sci USA, 1999, 96(24): 13910-13913.
[13] Worobey M, Holmes EC. Evolutionary aspects of recombination in RNA viruses [J]. J Gen Virol, 1999, 80 (Pt 10): 2535-2543.
[14] Sierra S, Dávila M, Lowenstein PR, Domingo E. Response of foot-and-mouth disease virus to increased mutagenesis: influence of viral load and fitness in loss of infectivity [J]. J Virol, 2000, 74(18): 8316-8323.
[15] Tong L, Lin L, Zhao W, Wang B, Wu S, Liu H, Zhong X, Cui Y, Gu H, Zhang F, Zhong Z. Destabilization of coxsackievirus B3 genome integrated with enhanced green fluorescent protein gene [J]. Intervirology,2011,54(5):268-275.
