二醋酸纖維素接枝聚甲基丙烯酸甲酯的合成與表征
2012-04-09 09:15:00丁建明董岸杰張建華鄧聯東
化學工業與工程 2012年4期
李 寧,丁建明,董岸杰,張建華,鄧聯東
(天津大學化工學院,天津 300072)
從資源的利用和環境保護來考慮,以天然高分子纖維素為基本原料,有可能開發出新型的生物降解高分子材料。二醋酸纖維素是纖維素衍生物中最早進行商品化生產的纖維素有機酸酯,用作煙用濾材和透析材料等[1-2]。由于該材料的流動溫度高以及較高的結晶度導致的加工分解等問題限制了其進一步的應用。接枝聚合作為材料改性的一種手段,已經有了很多研究。但是聚合之后較寬的相對分子質量分布又影響了材料原有的性能[3-5]。原子轉移自由基聚合作為接枝的一種方法具有反應速度快、反應溫度適中、適用單體范圍廣、相對相對分子質量可控等優點[6-8]。由于聚甲基丙烯酸甲酯的分子鏈較柔軟,韌性好,及其較好的生物相容性和透光性,能賦予共聚物優良的性質。本研究通過原子轉移自由基聚合技術將聚甲基丙烯酸甲脂引入到二醋酸纖維素的骨架上合成接枝型共聚物,并對產物的性能進行了表征。
1 試驗部分
1.1 原料
二醋酸纖維素(CDA,Mn=54 000,取代度為2.45),沈陽藥大制劑新技術有限公司;溴化鈉、五水合硫酸銅、無水亞硫酸鈉、濃硫酸、冰乙酸、乙醇、丙酮、甲醇、氫氧化鈉、三乙胺、1,4-二氧六環、四氫呋喃(THF),分析純,天津市江天化工技術有限公司;溴化亞銅(CuBr)、2-溴異丁酰溴、五甲基二乙烯三胺(PMDETA)和甲基丙烯酸甲酯(MMA),分析純,上海晶純試劑有限公司。
1.2 大分子引發劑CDA-Br的制備
在250 mL的燒瓶中加入5.0 g CDA用50 mL丙酮溶解,溶解完成后通氮氣,然后在冰浴條件下加入4.0 mL三乙胺,最后在恒壓條件下逐滴加入10.0 mL的2-溴異丁酰溴和10 mL丙酮,冰浴條件下反應1 h后,然后室溫避光反應24 h。再加入5 mL丙酮稀釋,過濾除鹽。在V(甲醇)∶V(水)為8∶2的混合溶液中沉淀并多次洗滌,最后再用甲醇洗滌,減壓抽濾,產物置于40 ℃真空烘箱中干燥24 h,得到CDA-Br。
1.3 CDA-g-PMMA接枝共聚物的合成
精確稱取0.17 g CDA-Br用2.0 mL的1,4-二氧六環在封閉反應管中超聲溶解,溶解后加入8 mL MMA和0.05 mL的PMDETA,抽真空、通氮氣和凍融循環3次,然后加入0.04 g的CuBr抽真空-通氮氣、凍融循環3次,溶解后放入70 ℃恒溫油浴中反應4 h(6和8 h),待反應結束后用30 mL的THF稀釋,然后在V(甲醇)∶V(水)為8∶2的溶液中沉淀并多次洗滌,最后用甲醇洗滌,減壓過濾,產物置于40 ℃的真空烘箱中干燥24 h,即得到所需產物。反應方程式如圖1所示。
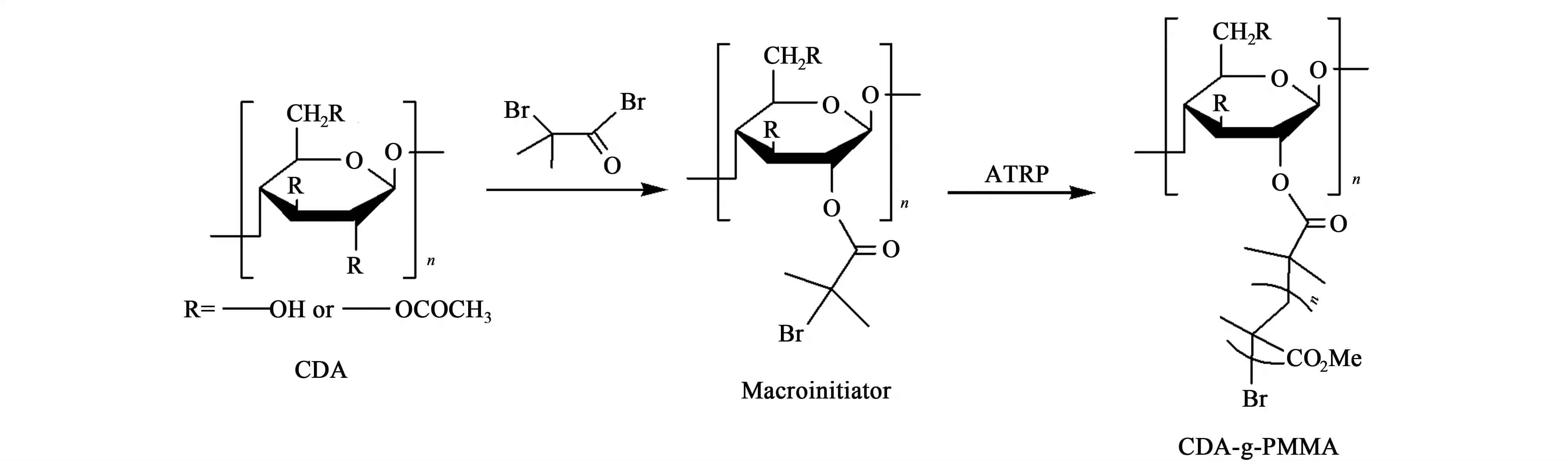
圖1 CDA-g-PMMA的合成路線Fig.1 Sythetic route of CDA-g-PMMA
1.4 CDA-g-PMMA的表征
1.4.1FT-IR分析
紅外光譜(IR),美國BIO-RAD FTS300紅外光譜儀,KBr壓片,400~4 000 cm-1。根據IR圖譜中相關峰的位置及峰形分析,確定樣品的結構和組成。
1.4.21H-NMR分析
采用Varian Unity-Plus 400型核磁共振光譜儀,以氘代丙酮為溶劑,測定CDA-g-PMMA的核磁光譜。根據譜圖中相關峰的位置及面積,確定CDA-g-PMMA的結構組成。
1.4.3DSC分析
CDA-g-PMMA的熱性能利用德國Netzsch公司204F1型動態熱流式差示掃描量熱計在0~250 ℃范圍內測試,升溫速率為10 ℃/min。
1.4.4XRD分析
CDA-g-PMMA的結晶性用德國Bruker公司D8 FOCUS型X-射線衍射儀,使用Cu_Kα射線,λ=0.154 05 nm,2θ=2~40°。
1.4.5凝膠滲透色譜(GPC)試驗
采用美國Agilent公司的Agilent1100型凝膠滲透色譜(GPC)-光散射聯用測定聚合物的相對相對分子質量和多分散系數,測定溫度為25 ℃,THF為溶劑,控制流速為1 mL/min。
1.4.6拉伸試驗
將CDA和CDA-g-PMMA材料制成長5.5 cm,寬2 cm,厚度為(0.07±0.02) mm的薄片,利用萬能材料測試儀(M350-Testometric)進行拉伸試驗。
2 結果與討論
2.1 CDA-Br的紅外表征結果
CDA與CDA-Br的紅外譜圖如圖2所示。

圖2 CDA和CDA-Br的紅外光譜Fig.2 IR spectra of CDA and CDA-Br

2.2 1H-NMR分析結果
CDA-g-PMMA的核磁譜圖如圖3所示。
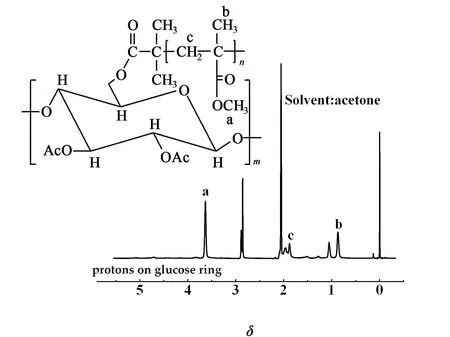
圖3 CDA-g-PMMA的核磁共振譜圖Fig.3 1H-NMR spectrum of CDA-g-PMMA
δ為2.05和2.85處出現的化學位移峰分別為溶劑殘余峰和殘留水峰,δ為 0.84~1.12是甲基(Hb)的質子化學位移峰;δ為1.84~2.04處是—CH2—(Hc)的質子化學位移峰;δ為 3.64處出現的是醋酸基甲基(Ha)的質子化學位移峰,δ為3.60~5.10是纖維素環上C—H和C6上亞甲基的化學位移峰。由此得出,成功合成了CDA-g-PMMA的接枝共聚物。
2.3 DSC分析結果
接枝共聚物的熱性能通過DSC進行分析,見圖4。
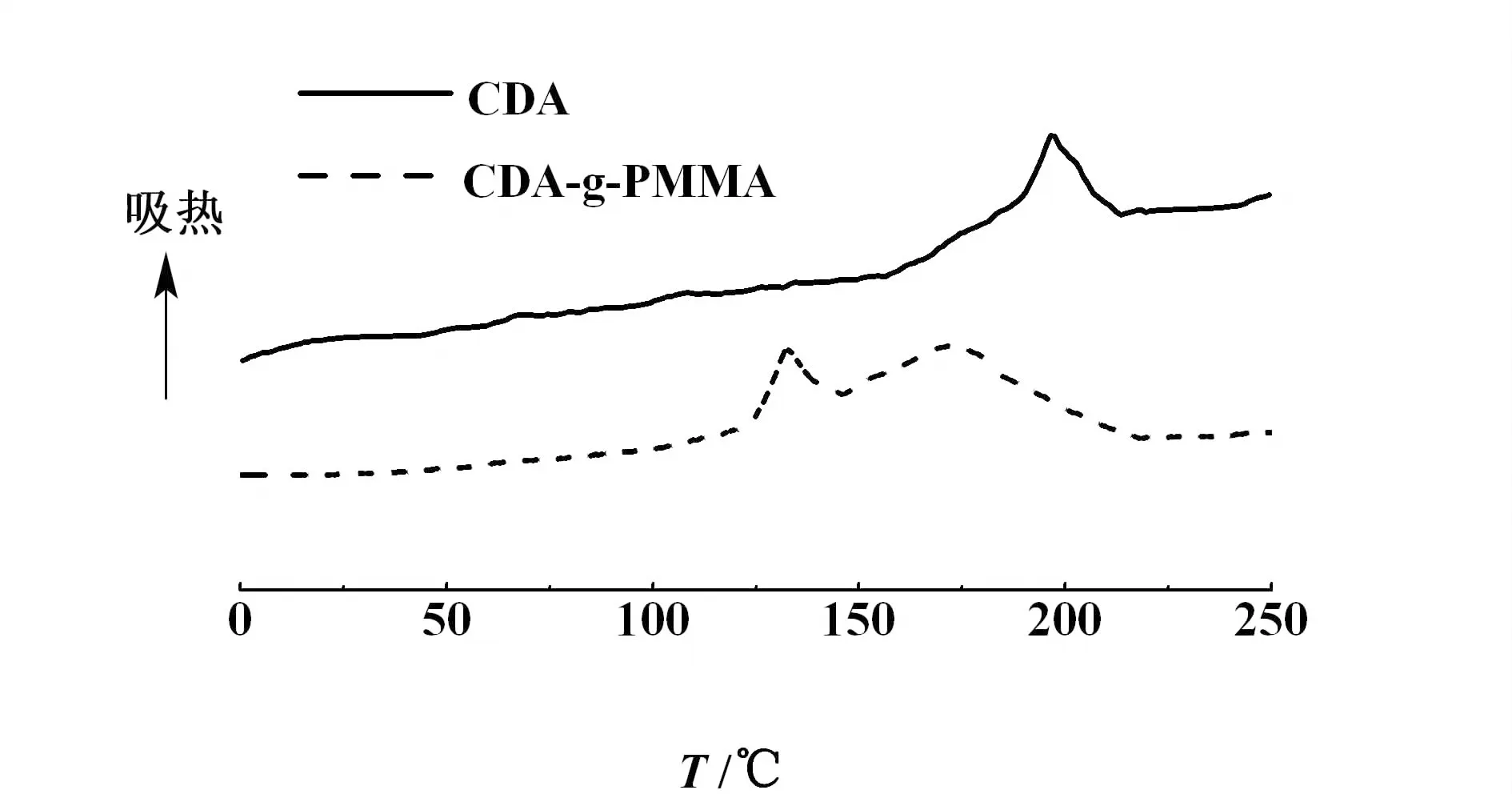
圖4 CDA和CDA-g-PMMA的DSC曲線Fig.4 DSC curves of CDA and CDA-g-PMMA
由圖4可以看出,CDA-g-PMMA在130 ℃出現的是PMMA的熔融峰,而在175 ℃出現的是CDA的熔融峰,而接枝前CDA的熔融峰出現在200 ℃左右,接枝聚合物的熔點降低,這說明PMMA鏈段的引入對CDA的結晶產生了一定程度的影響,使分子間的相互作用力減弱,分子鏈在熱作用下能夠較為自由的運動,這也說明CDA-g-PMMA的熱塑性得到一定改善。
2.4 XRD分析結果
圖5為CDA和CDA-g-PMMA的X-射線衍射曲線。
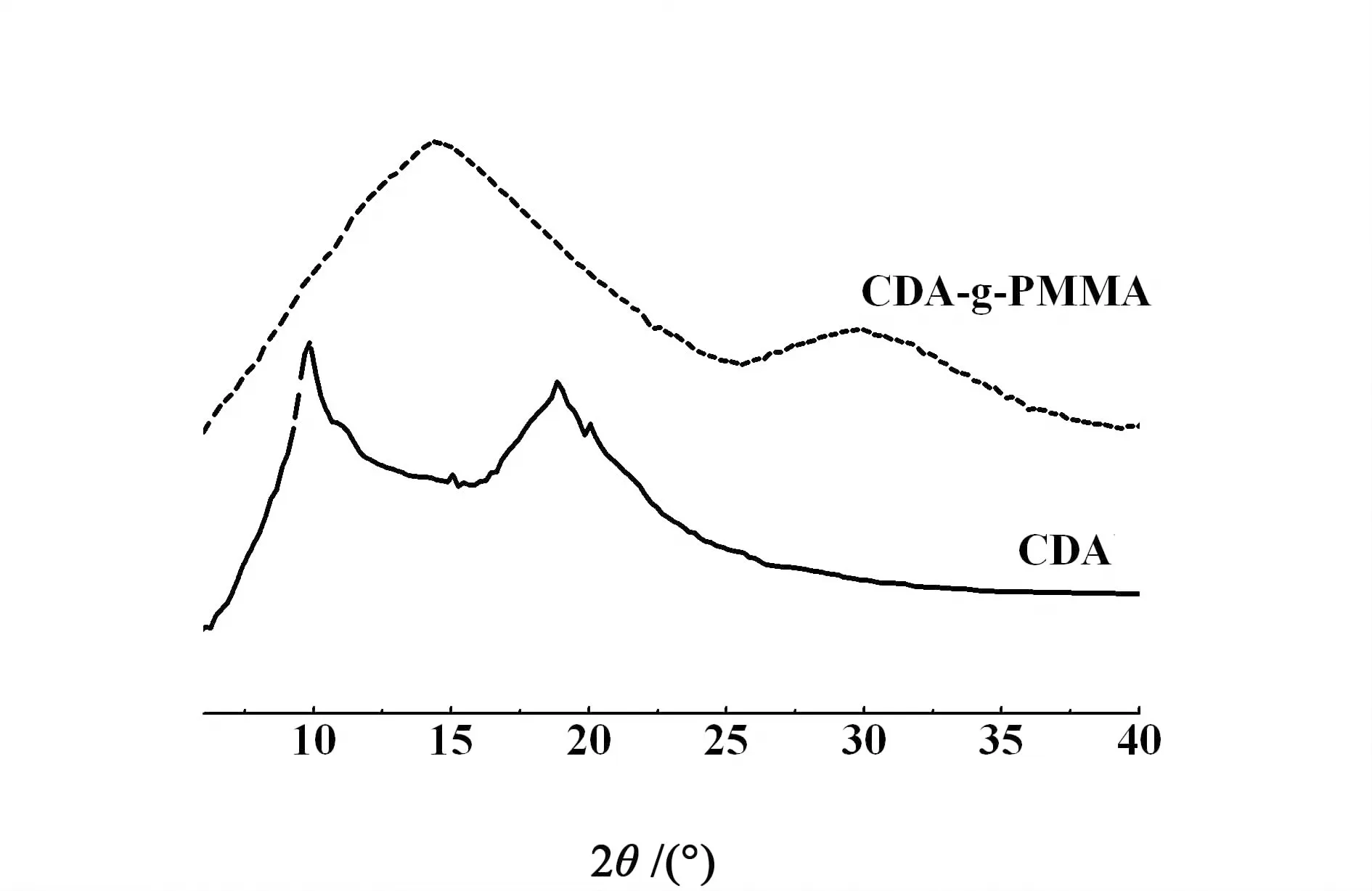
圖5 CDA和CDA-g-PMMA的XRD圖Fig.5 XRD pattens of CDA and CDA-g-PMMA
從圖5中知道CDA在2θ為10和20 °左右出現了2個結晶峰,其結晶度為54%。當接枝PMMA后結晶性發生很大變化,CDA-g-PMMA中只有2個彌散漫衍射峰,這是因為接枝PMMA后,CDA上的羥基被取代,氫鍵消失,分子間的作用力減弱,分子鏈自由的移動能力增強所致,這也進一步證明了上述的觀點。
2.5 GPC分析結果
圖6為CDA-g-PMMA和CDA的GPC曲線,所有GPC淋洗曲線均呈單峰,接枝后的峰位置向左偏移,相對分子質量變大,這就說明合成的產物不是CDA和MMA的均聚物,而是CDA-g-PMMA的接枝共聚物。從表1可知,聚合物的相對分子質量隨反應時間的延長是成線性增長的,而且相對分子質量分布系數較小,這也說明ATRP方法在該接枝聚合中起到了應有的作用。

圖6 CDA和CDA-g-PMMA的GPC曲線Fig.6 GPC curves of CDA and CDA-g-PMMA

表1 反應時間對CDA-g-MMA的影響Table 1 Effect of reaction time on CDA-g-PMMA
a:反應投入為n(MMA):n(CDA-Br),b:通過GPC測得。
2.6 拉伸試驗結果
從圖7中我們清晰地看到改性之后CDA-g-PMMA的斷裂伸長率明顯大于CDA,分別為3.9%和2.8%。CDA的應力-應變曲線較平緩,出現該現象的本質是分子發生取向的結晶,拉伸一定程度時應力幾乎不變而式樣變形很大,而改性后則不然。結果證明改性后材料的韌性得到改善。
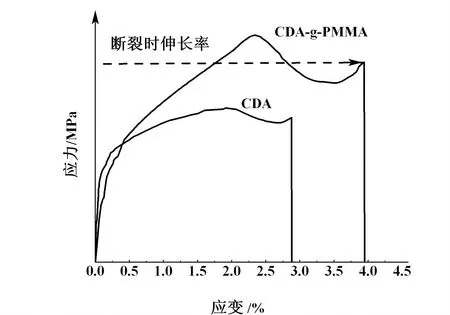
圖7 CDA和CDA-g-PMMA的應力-應變曲線Fig.7 Stress-strain curves of CDA and CDA-g-PMMA
3 結論
以CDA為接枝骨架,首先將未被酯化的羥基酰溴化,再利用ATRP方法接枝PMMA,成功地合成了CDA-g-PMMA。改變聚合時間可以合成不同相對分子質量的接枝共聚物,說明ATRP技術適用于天然大分子的改性,得到的聚合物相對分子質量可控、相對分子質量分布較窄。PMMA鏈段的引入降低了熔融溫度和結晶度,增強了材料的韌性。
參考文獻:
[1]葉代勇,黃洪,傅和青,等.纖維素化學研究進展[J].化工學報,2006,5(8):1 782-1 791
[2]RODRIGUES F G,MONTEIRO D S,MEIRELES C S,etal.Synthesis and characterization of cellulose acetate produced from recycled newspaper[J].Carbohydrate Polymers,2008,73:74-82
[3]鄧文鍵,關克田,莊旭品,等.纖維素熱塑改性研究進展[J].化工時刊,2010,24(12):44-47
[4]楊莉燕,譚惠民.二醋酸纖維素接枝聚己內酯的制備及性能研究[J].功能材料,2007,38:3 530-3 532
[5]王東山,黃勇,沈家瑞.二醋酸纖維素與聚乙二醇單甲醚接枝共聚物的合成與表征[J].纖維素科學與技術,2001,9(3):19-26
[6]嚴微,彭慧,程時遠,等.原子轉移自由基聚合研究進展[J].膠體與聚合物,2005,23(1):38-40
[7]VLCEK P,JANATA M,LATALOVA P,etal.Controlled grafting of cellulose diacetate[J].Polymer,2006,47:2 587-2 595
[8]MATYJASZEWSKI K,XIA J.Atom transfer radical polymerization[J].Chemical Reviews,2001,101(9):2 921-2 990
猜你喜歡
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
中國塑料(2016年12期)2016-06-15 20:30:07
汽車觀察(2016年3期)2016-02-28 13:16:26
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
