動態交聯聚丙烯/乙烯-醋酸乙烯酯共聚物結晶行為
2012-06-11 03:22:40江學良范一泓吳天驥曾溢宇
武漢工程大學學報 2012年11期
江學良,李 凡,范一泓,任 軍,吳天驥,曾溢宇
(武漢工程大學材料科學與工程學院,湖北 武漢 430074)
0 引 言
聚合物共混物的性能不僅與其配比有關系,還受其形態結構等等方面的影響[1-2].而通常共混物的形態結構則由下述因素決定:配比、結晶行為、黏度以及工藝條件等.因此,研究共混物這些方面的參數對系統地研究新材料時有著重要的意義.
結晶性聚合物在其成型加工過程中會發生結晶行為,這種現象會直接影響到聚合物制品的使用性能.所以,研究聚合物結晶過程中的各項參數并了解這些參數的影響條件,在制定配方和選擇合適的工藝條件上有著非常重要的作用.
國內外的學者和單位對純聚丙烯及共混物中聚丙烯的結晶行為已進行了大量的研究[3-5].Li等[3]研究了聚丙烯(PP)/聚碳酸酯(PC)中PP的結晶行為,發現PC在體系中起到PP成核劑的作用,促進了PP的結晶.Zhang等[4]通過研究,發現乙烯-辛烯共聚物(POE)加入共混體系中也能夠促進PP的結晶.Xu[5]等則系統地研究了不同成核劑對PP成核作用的影響.
本研究成功地將動態交聯技術應用到聚丙烯(PP)/乙烯-醋酸乙烯酯共聚物(EVA)共混體系中,制備了一種動態交聯PP/EVA共混物.動態交聯PP/EVA共混物的力學性能和加工性能直接與PP的結晶行為有關.研究動態交聯PP/EVA共混物中PP的結晶過程有利于材料配方優化和成型工藝條件的選擇.迄今為止,有關動態交聯PP/EVA共混物中PP的結晶行為研究,還少見文獻報道.在本研究中,研究了動態交聯PP/EVA共混物中PP的非等溫結晶動力學,同時還考察了共混物中PP的晶體結構.
1 實驗部分
1.1 主要原料
聚丙烯(PP),牌號T30S,中國石化天然氣股份有限公司生產;乙烯-醋酸乙烯酯共聚物(EVA),VA質量分數為28%,揚子石化有限公司生產;過氧化二異丙苯(DCP),國藥集團生產;氧化鎂(MgO),山東魯華化工有限公司生產.
1.2 共混物的制備
共混前,PP、EVA于80 ℃真空干燥約10 h.動態交聯PP/EVA共混物在南京誠盟化機械有限公司SHJ-36型雙螺桿共混擠出機上制得,共混溫度為190 ℃,轉速為180 r/min.先將PP、EVA、DCP等按照一定比例置于高速混合機中充分混合均勻,然后加入雙螺桿擠出機擠出造粒成型,干燥后待用.
1.3 性能測試
1.3.1 DSC分析 樣品的非等溫結晶過程測試在差示掃描量熱儀(美國TA公司,Q10型)上進行,儀器經銦標校正,實驗氣氛為N2,氣體流速為20 mL/s.每次取樣品5~10 mg.將試樣以20 ℃/min升溫至210 ℃,恒溫6 min.然后以設定的降溫速率勻速降溫至40 ℃,記錄結晶過程中的熱焓隨著溫度的變化.
1.3.2 X射線衍射分析 將試樣壓制成2 mm厚片,采用日本理學D/MAX-Ⅲ型X射線衍射儀測定樣品的晶型.在管電流40 mA,管電壓40 kV的CuKα(發射波長1.540 6×10-10m)射線條件下,以4°/min的掃描速度對2θ=5~60°的范圍進行掃描.
2 結果與討論
2.1 非等溫結晶行為
圖1是PP和動態交聯PP/EVA共混物在不同降溫速率下的非等溫結晶DSC曲線.而結晶過程參數見表1.
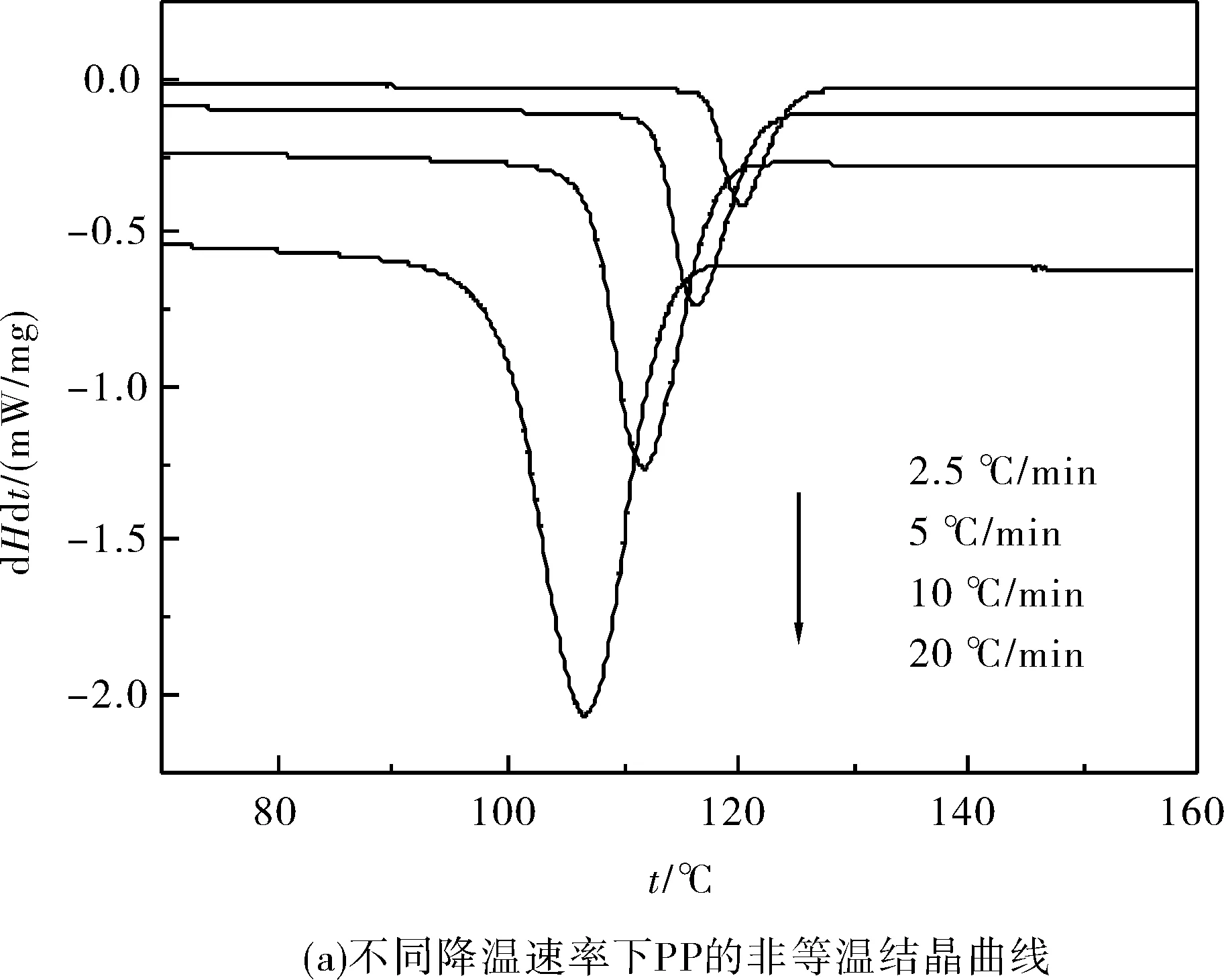
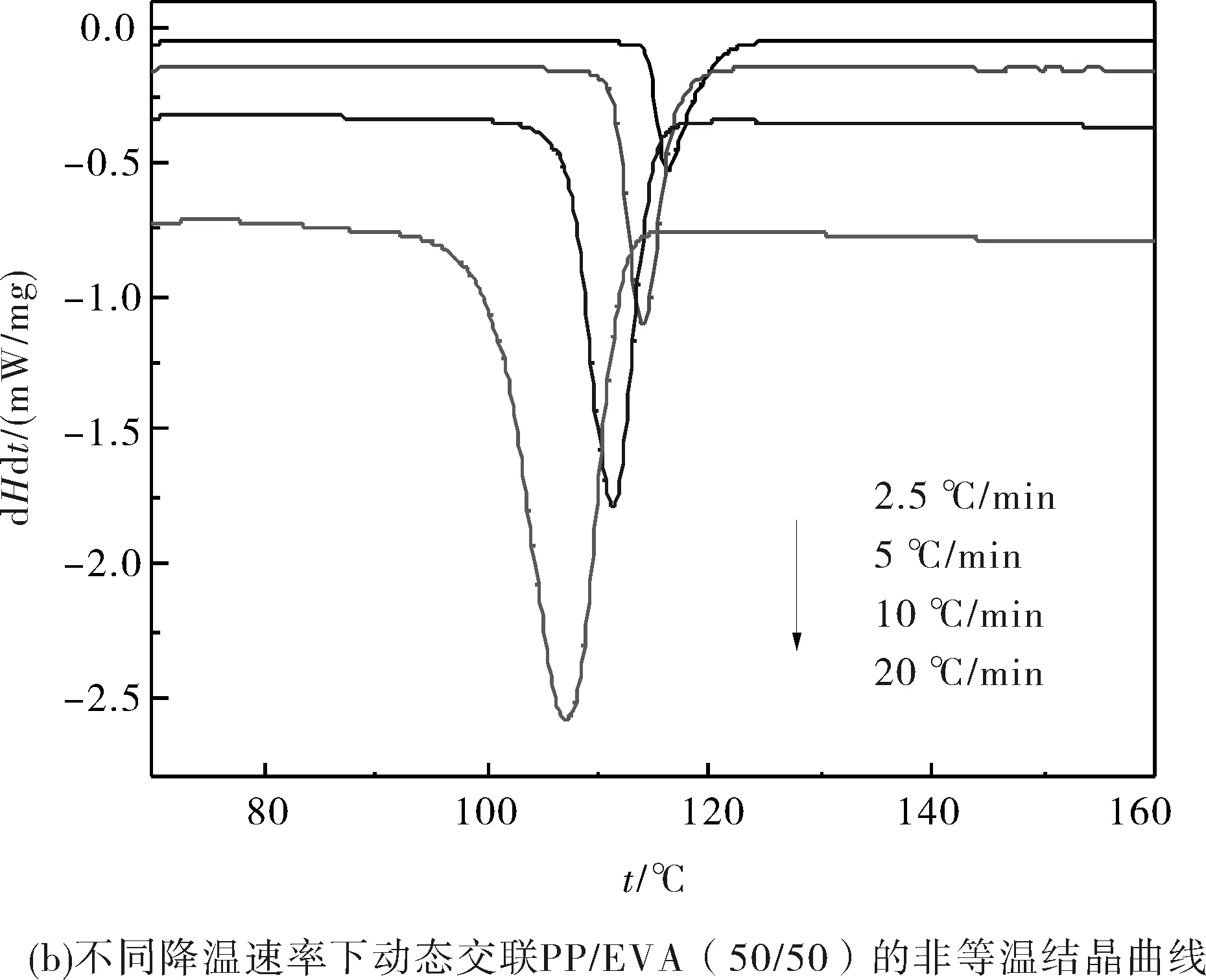
圖1 不同降溫速率下PP和動態交聯PP/EVA共混物的非等溫結晶DSC曲線Fig.1 DSC curves of nonisothermal crystallization at different cooling rate
從表1可知,隨著降溫速率的增加,材料的結晶放熱峰變寬并且向著低溫方向移動;試樣的結晶初始溫度(Ti)、結晶結束溫度(Te)和結晶峰溫度(Tp)也都向著低溫方向移動.完成整個結晶過程所用的時間(tc)隨著降溫速率的提高而縮短.
在相同的降溫速率下,PP/EVA共混物及動態交聯PP/EVA的Ti較PP低且完成整個結晶過程的時間(tc)明顯縮短.這表明共混物中的EVA顆粒起到了異相成核的作用,促進了PP的結晶.對比PP/EVA和動態交聯PP/EVA的各項數據,發現動態交聯以后,共混物的Ti、Te和Tp在純PP/EVA共混體系的基礎上均發生了下降,并且完成結晶過程的時間tc略有下降.這說明動態交聯EVA強化了EVA顆粒的異相成核作用.
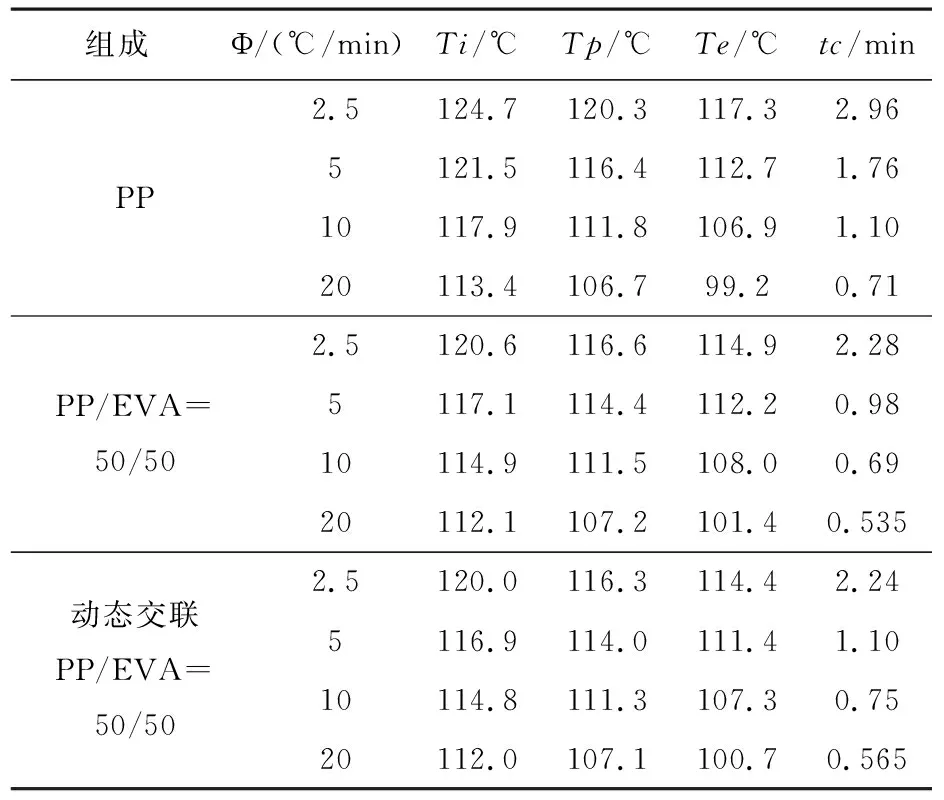
表1 不同降溫速率下PP和PP/EVA共混物的非等溫結晶過程參數Table 1 Parameters for nonisothermal crystallization of PP and PP/EVA blends at different cooling rate
圖2為PP和PP共混物中的PP在不同降溫速率下的相對結晶度隨著溫度變化的曲線圖.從圖中可知,隨著降溫速率的提高,各組共混物中的PP達到同一相對結晶度X(T)時所需的溫度明顯降低;而在同一降溫速率下,PP共混物中的PP達到某一結晶度時所需要的溫度要高于純PP所需的溫度.
2.2 Ozawa法計算非等溫結晶動力學參數
有關聚合物非等溫結晶動力學的研究有多種模型,其中Avrami[6-7]方程是最常用的模型之一.但是因為其沒有考慮到連續降溫對結晶造成的影響,所以用Avrami方程來分析非等溫結晶過程得不到良好的線性關系.Ozawa[8]則考慮了這一重要因素,在Avrami方程的基礎上得到了對非等溫結晶過程更為適用的結晶動力學方程.根據Ozawa模型,聚合物在一定溫度下某一降溫速率時的相對結晶度X(T)可由下式計算:
(1)
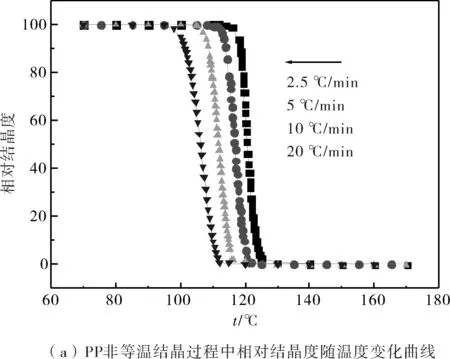
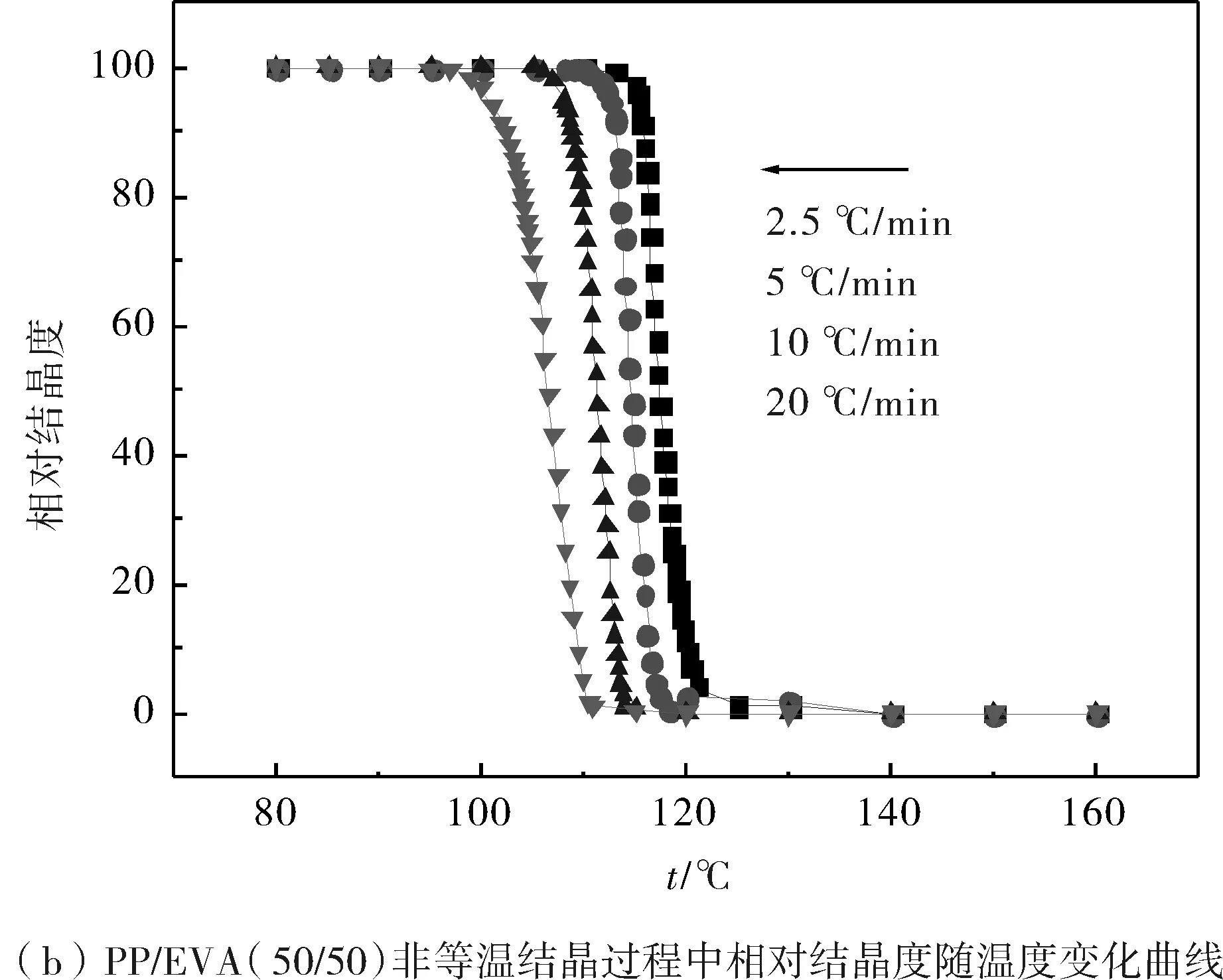
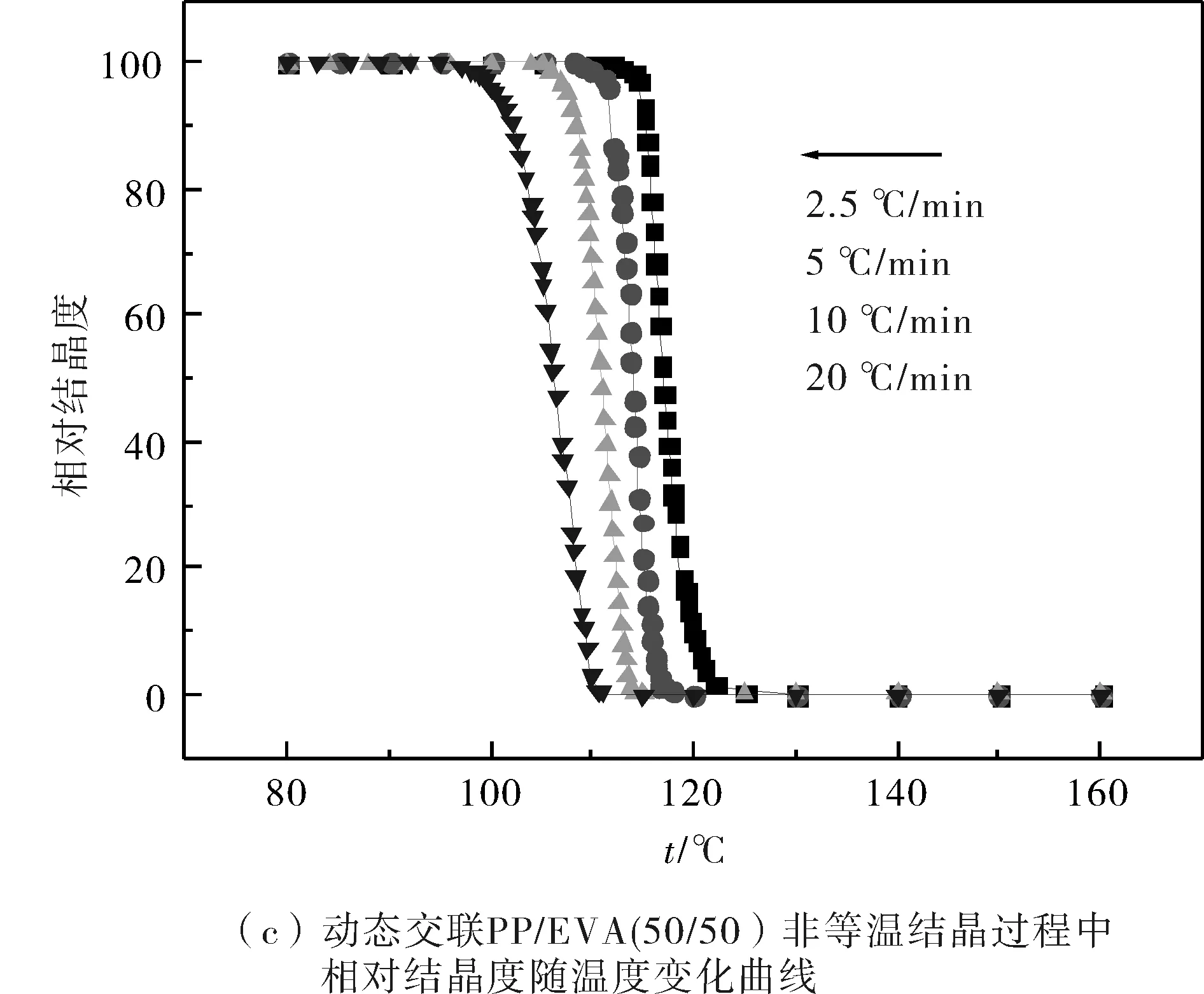
圖2 PP和不同PP/EVA共混物非等溫結晶過程中相對結晶度隨溫度的變化Fig.2 Development of relative crystallinity with temperature for nonisothermal crystallization of PP and PP/EVA blends
式中,X(T)是溫度為T時的相對結晶度,?是降溫速率,m是Ozawa指數,是與成核結晶機理及生長方式有關的常數.而K*(T)則是與成核速率、成核方式及晶核的生長速率等因素有關的參數,是溫度的函數.X(T)可以根據以下公式計算:

(2)
式中,QT和QT∞是在結晶溫度為T和結晶溫度無限高時釋放的熱量.dH/dT為熱量流率.
式(1)取對數后可得下式:
log[-In(1-X(T))]=logK*(T)-mlog?
(3)
從上式可以知道,在一定溫度下,以log[-In(1-X(T))]對log?作圖,所得的直線斜率為-m,截距則為logK*(T).
由圖3可知,結晶過程中在達到同一溫度時,試樣的結晶度的對數與降溫速率的對數之間存在較好的線性關系,這說明Ozawa模型是能夠有效地處理非等溫結晶過程的結晶動力學的.綜合所得的非等溫結晶參數見表2.
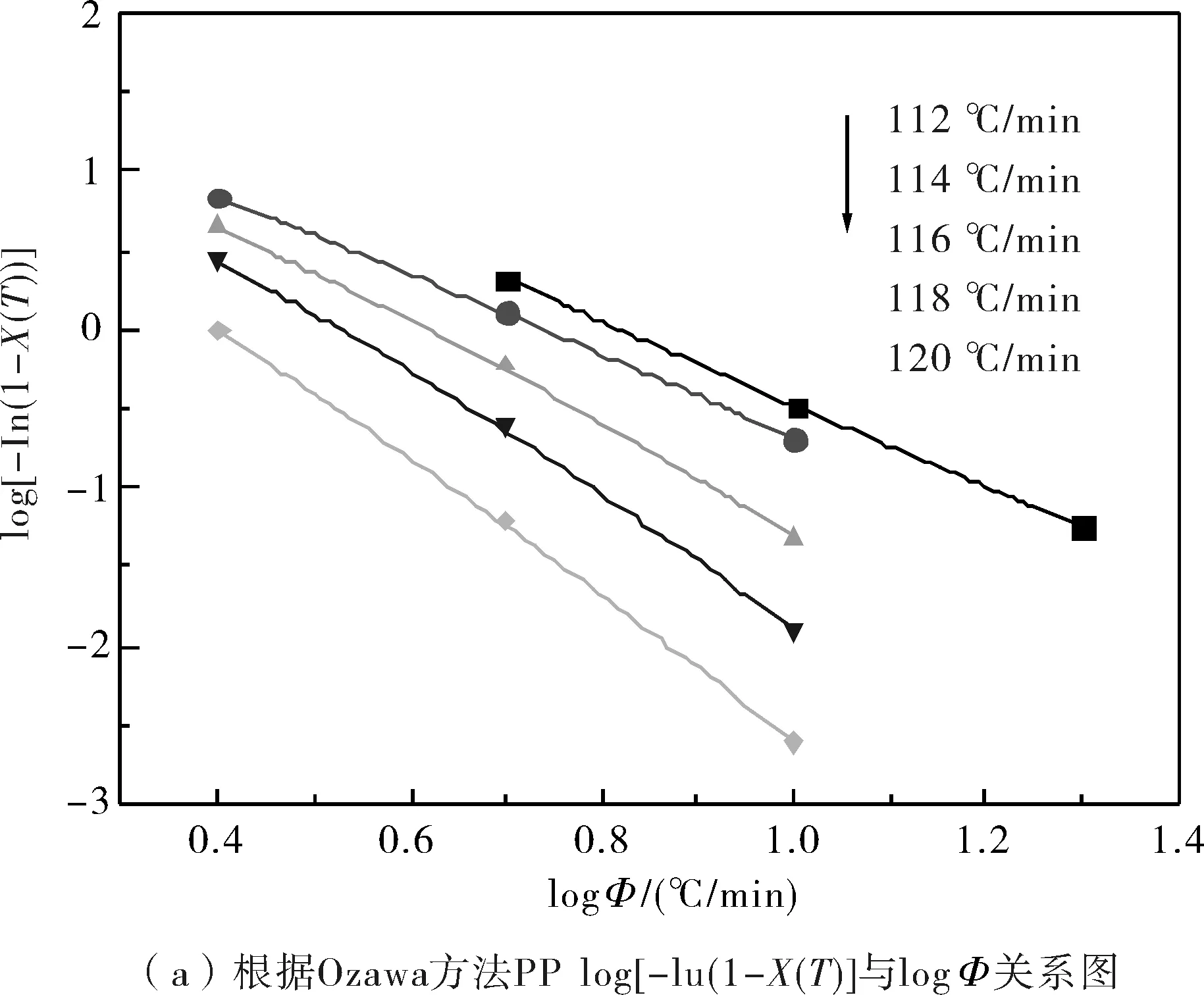
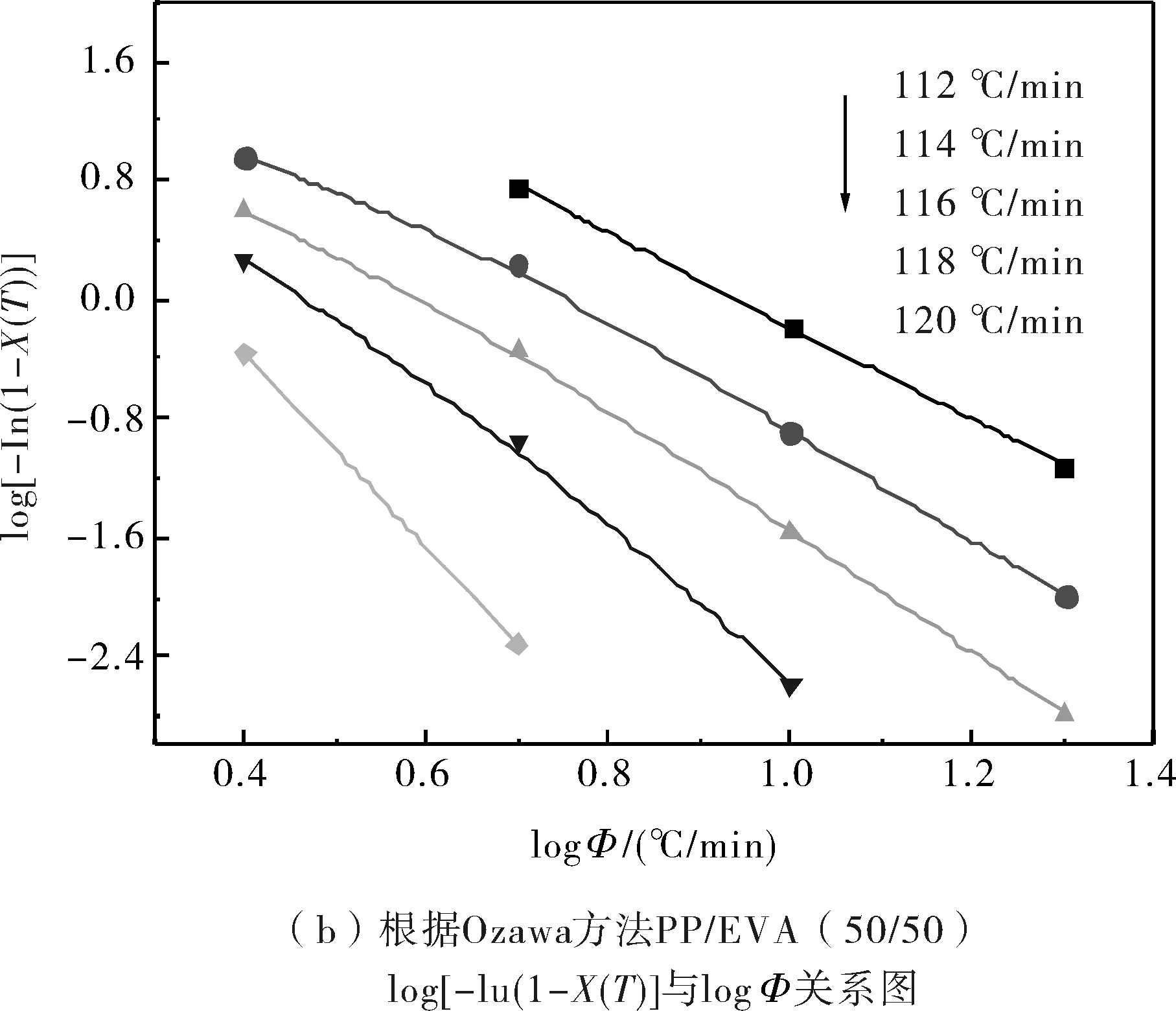
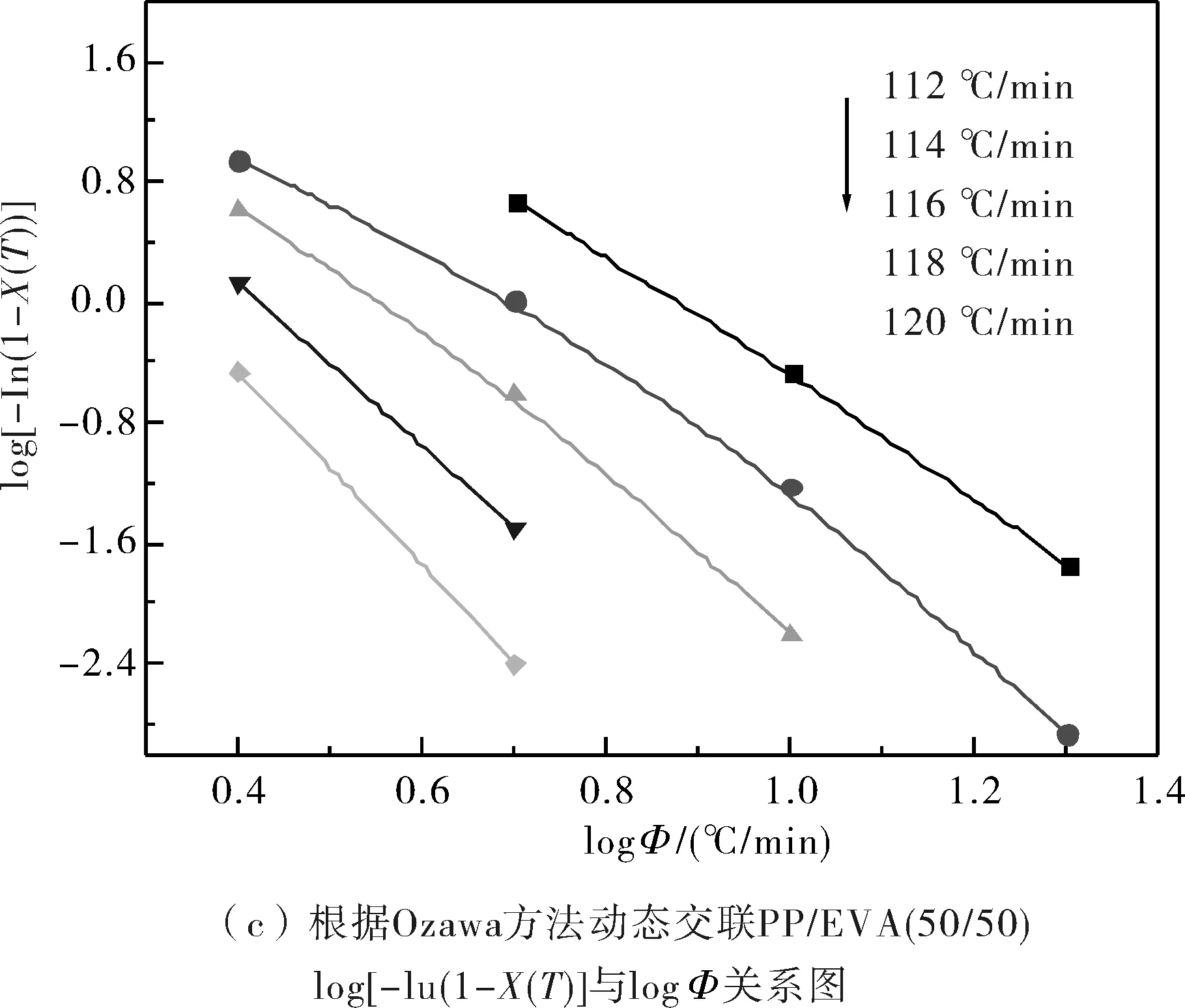
圖3 根據Ozawa方法log[-In(1-X(T))]與log?關系圖Fig.3 log[-In(1-X(T))] as a function of log? from Ozawa theory
從表2中可知,純PP的Ozawa指數m約為2~4,而PP/EVA共混物中的PP的Ozawa指數m約為3~7,動態交聯共混物中PP的m值較純PP/EVA共混物中PP的m值大.這表明共混物中的純EVA顆粒和動態交聯的EVA顆粒在PP的結晶過程中都能起到異相成核劑的作用,從而導致PP結晶的成核與生長等方面都發生了改變.
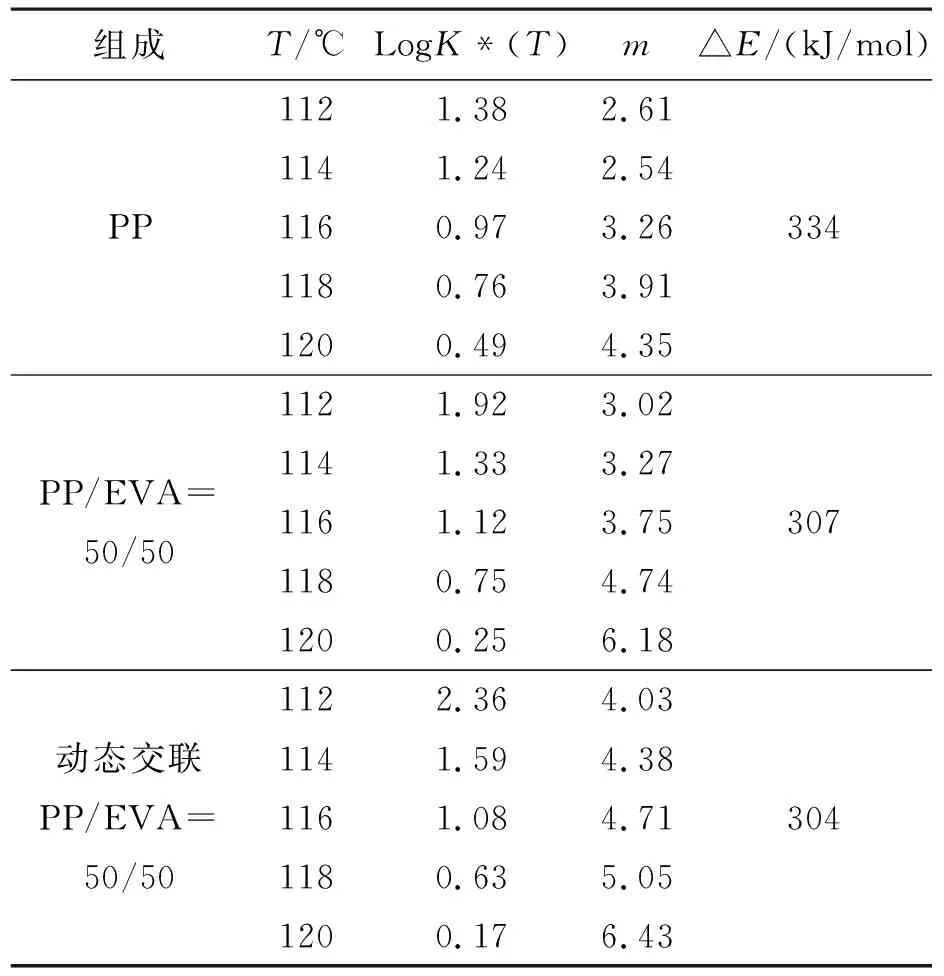
表2 根據Ozawa方法獲得的非等溫結晶動力學參數Table 2 Parameters of nonisothermal crystallization from Ozawa theory
Kissinger[9]研究了不同降溫速率對結晶過程的影響,提出了計算非等溫結晶過程活化能ΔE的公式:
(4)
式中,R是氣體常數,?是降溫速率,Tp是結晶峰溫度.對公式(4)進行積分整理,可以得到活化能的計算公式:
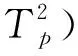
(5)
將表1的數據代入公式(5)可算出非等溫結晶過程的活化能見表2.從表中可知兩種PP/EVA共混物的活化能均小于純PP結晶的活化能,這表明EVA顆粒與經動態交聯的EVA顆粒在體系中都可以促進PP的非等溫結晶過程.另外,動態交聯PP/EVA共混物中的PP的活化能較之于簡單共混物中的PP略低.
2.3 X射線衍射分析
圖4是PP和不同共混體系的X射線衍射圖譜,從圖中可知,PP和不同體系共混物在衍射角(2θ)為14°、17.1°、18.7°、22.4°處分別有一個衍射峰,這是PP典型的α晶型表現.實驗結果說明,EVA的加入和動態交聯都不會改變共混體系中PP的晶體結構.
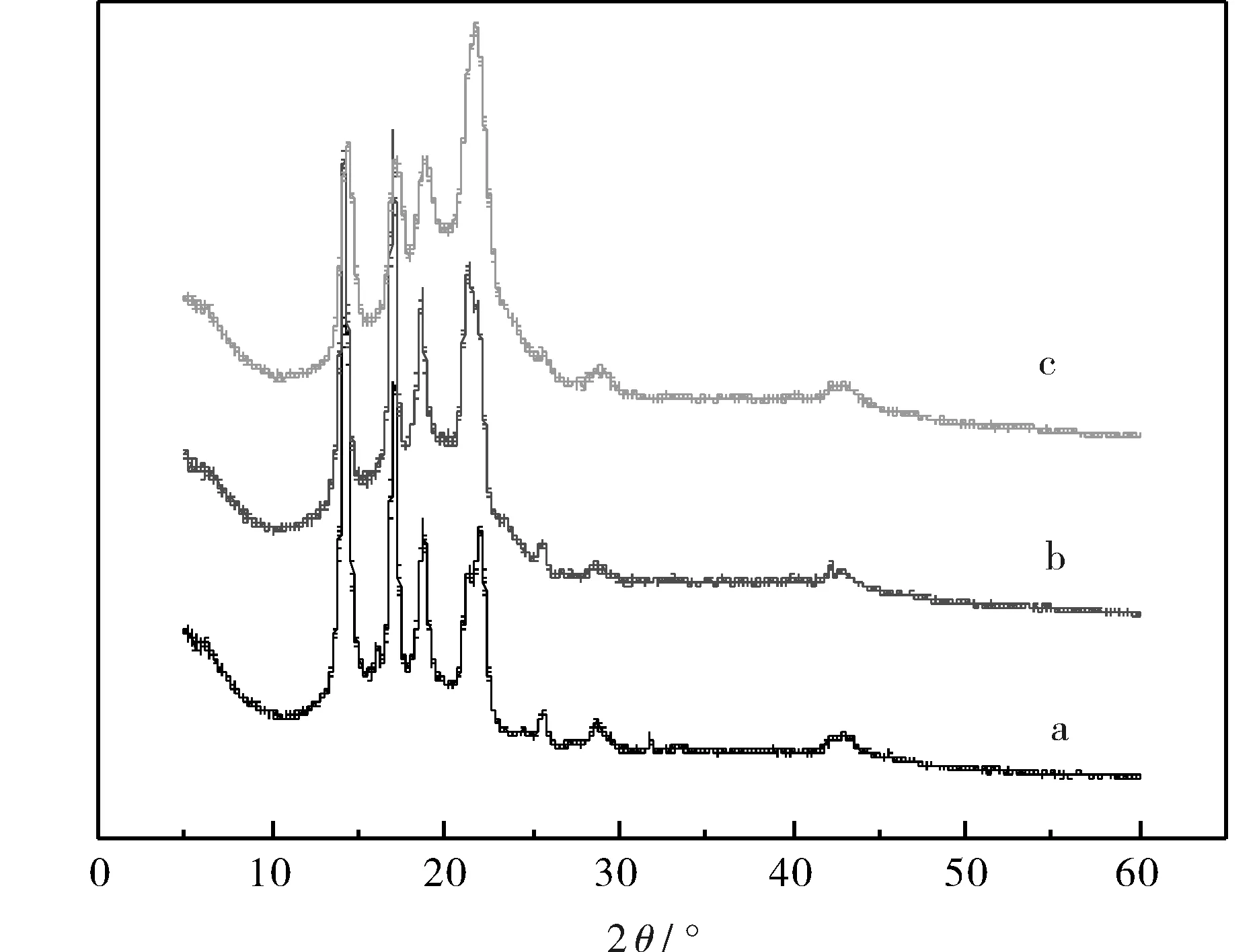
圖4 PP和不同體系PP/EVA共混物的X射線衍射圖譜Fig.4 X-ray diffraction patterns of PP and different systems of PP/EVA blends注: (a)PP;(b)PP/EVA=50/50;(c)動態交聯PP/EVA=50/50.
3 結 語
a. 差示量熱掃描(DSC)研究表明,動態交聯PP/EVA共混物中EVA起到異相成核作用,促進了PP的結晶.
b. Ozawa方程適合于描述PP和PP/EVA共混物的非等溫結晶動力學.在同一溫度下,PP/EVA和PP/EVA/DCP共混物中的PP結晶速率大于純PP的結晶速率.根據Kissinger方法計算得知,PP/EVA共混物和動態交聯PP/EVA共混物中的PP結晶活化能(ΔE)均小于純PP的ΔE,且后者略小于前者.
c. X射線衍射分析發現,共混和動態交聯都沒有改變共混物中PP的晶體結構,仍是α晶型.
參考文獻:
[1] Albano C, Gonzalez J, Lchazo M. Mechanical and morphological behavior of polyolefin blends in the presence of CaCO3[J]. Composite Structure, 2000, 48: 49-58.
[2] Chun Y S, Jung H C, Han M S, et al. Crystalliza-tion behavior and rheological properties of polycar-bonate and polypropylene blends[J]. Polymer Eng-ineering and Science, 1999, 39: 2304-2312.
[3] Li C Q, Zhang Y, Zhang Y X. Crystallization beha-vior of polypropylene/polycarbonate blends[J]. Po-lymer Testing, 2002, 21(8): 919-926.
[4] Zhang X F, Xie F, Peng Z L, et al. Effect of nuclea-ting agent on the structure and properties of polypr-opylene/poly(ethylene-octene) blends[J]. European Polymer Journal, 2002, 38: 1-6.
[5] Xu T, Lei H, Xie C S. The effect of nucleating agent on the crystalline morphology of polypropylene[J]. Materials and Design, 2003, 24(3): 227-230.
[6] Avrami M J. Kinetics of phase change-I. General theory[J]. Journal of Chemistry and Physics, 1939(7): 1103-1109.
[7] Avrami M. Kinetics of phase change-III. Granula-tion, phase change and microstructure[J]. Journal of Chemistry and Physics, 1941(9): 117-124.
[8] Li J, Zhou C X, Wang G. Isothermal and noniso-thermal crys-tallization kinetics of elastomeric polyp-ropylene[J]. Polymer Testing, 2002, 21: 583-589.
[9] Kissinger H E. Variation of peak temperature with heating rate in different thermal analysis[J]. J Research Natl Bur Standards, 1956, 57: 217-221.
