Akt-eNOS-NO信號途徑介導了Ang-1和Ang-2對大鼠失血性休克早期血管高反應性的調節作用*
2012-11-06 05:46:02楊光明劉良明
中國病理生理雜志 2012年9期
徐 競, 藍 丹, 楊光明, 李 濤, 劉良明
(第三軍醫大學大坪醫院野戰外科研究所二室,創傷、燒傷與復合傷國家重點實驗室,重慶 400042)
1000-4718(2012)09-1554-05
2011-10-25
2012-07-16
國家自然科學基金資助項目(No.30801189);重慶市自然科學基金資助項目(No. 2008BB5103)
△通訊作者 Tel: 023-68757421;E-mail:Liuliangming2002@yahoo.com
Akt-eNOS-NO信號途徑介導了Ang-1和Ang-2對大鼠失血性休克早期血管高反應性的調節作用*
徐 競, 藍 丹, 楊光明, 李 濤, 劉良明△
(第三軍醫大學大坪醫院野戰外科研究所二室,創傷、燒傷與復合傷國家重點實驗室,重慶 400042)
目的觀察內皮型一氧化氮合酶(endothelial nitric oxide synthase, eNOS)在血管生成素1(angiopoietin-1, Ang-1)和血管生成素2(angiopoietin-2, Ang-2)調節失血性休克大鼠血管反應性雙相變化中的作用及其機制。方法采用Western blotting技術觀察失血性休克后不同時點腸系膜上動脈(superior mesenteric artery,SMA)中eNOS蛋白表達變化,采用離體微血管環張力測定技術觀察eNOS抑制劑對Ang-1和Ang-2調節缺氧早期和晚期血管反應性作用的影響,并觀察給予Ang-1、Ang-2以及Tie-2、Akt、p38 MAPK、ERK抑制劑后缺氧血管內皮細胞(vascular endothelial cells, VECs)和血管平滑肌細胞(vascular smooth muscle cells, VSMCs)混合培養物中eNOS蛋白表達和培養上清一氧化氮(nitric oxide, NO)含量的影響。結果(1) eNOS在正常SMA中表達很低,失血性休克后逐漸增高,休克10 min、30 min、1 h、2 h和4 h時分別增高至正常對照的1.95、2.10、3.01、3.42和3.57倍(P<0.01)。(2)eNOS抑制劑可顯著抑制缺氧10 min的血管高反應性,去甲腎上腺素(NE)的Emax由13.479 mN降低至9.043 mN (P<0.01),也可以顯著抑制Ang-1對缺氧10 min血管高反應性的維持作用,NE的Emax由15.283 mN降低至11.219 mN (P<0.01),但不改變Ang-2降低缺氧10 min血管高反應性作用,也不改變缺氧4 h的血管低反應性和Ang-1、 Ang-2對缺氧4 h血管反應性的調節作用。(3)缺氧10 min的eNOS表達較正常對照增高,Ang-2和Tie-2、Akt抑制劑可抑制其增高(P<0.01),p38 MAPK、ERK抑制劑對其無顯著影響。(4)NO含量在缺氧10 min顯著增高,Ang-2和Tie-2、Akt、eNOS抑制劑可抑制其增高 (P<0.01),p38 MAPK、ERK抑制劑對其無顯著影響。結論失血性休克早期,Ang-1和Ang-2通過Akt-eNOS-NO途徑來調節血管高反應性。
失血性休克; 血管反應性; 雙相變化; 血管生成素1; 血管生成素2; 內皮型一氧化氮合酶
血管生成素1(angiopoietin-1, Ang-1)和血管生成素2(angiopoietin-2, Ang-2)屬于血管內皮細胞(vascular endothelia cells, VECs)特異的促血管新生因子家族——血管生成素家族,本實驗室前期研究發現,Ang-1和Ang-2在失血性休克后的腸系膜上動脈(superior mesenteric artery,SMA)中呈時間上的差異表達,并通過Tie-2受體參與了失血性休克血管反應性雙相變化的形成[1],蛋白激酶B(protein kinase B, PKB/Akt)、p38絲裂原活化蛋白激酶(p38 mitogen-activated protein kinase, p38 MAPK)、細胞外信號調節激酶(extracellular signal-regulated kinase, ERK)幾種分子參與了其調節過程,但具體的調節機制如何,目前尚不清楚。 根據基礎研究,Ang-1和Ang-2激活VECs上的Tie-2受體之后,可經磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase, PI3K)激活Akt,還可經過銜接蛋白Grb2活化p38 MAPK和ERK,再通過調節內皮型一氧化氮合酶(endothelial nitric oxide synthase, eNOS)產生一氧化氮(nitric oxide, NO)[2-3],而NO具有調節血管舒縮反應性的作用[4]。那么Ang-1和Ang-2是否通過Akt、p38 MAPK和ERK,調節eNOS產生NO,來調節失血性休克血管反應性的雙相變化? 本實驗采用Western blotting技術和離體微血管環張力測定技術,以大鼠SMA以及混合培養的VECs和血管平滑肌細胞(vascular smooth muscle cells, VSMCs)為研究對象,觀察失血性休克后不同時點SMA中eNOS蛋白表達變化,eNOS抑制劑對Ang-1和Ang-2調節缺氧早期和晚期血管反應性作用的影響,給予Ang-1、Ang-2以及Tie-2、Akt、p38 MAPK和ERK抑制劑后缺氧培養的VECs和VSMCs中eNOS蛋白表達和培養上清NO含量的變化,以探討eNOS在Ang-1和Ang-2調節休克血管反應性雙相變化中的作用及其信號途徑。
材 料 和 方 法
1動物和細胞準備
采用第三軍醫大學大坪醫院實驗動物中心清潔級SD大鼠,雌雄各半,體重200~230 g,戊巴比妥鈉(30 mg/kg大鼠體重)腹腔注射麻醉。
根據文獻分別進行VECs 和VSMCs原代細胞培養[5],取第3代至第5代的VECs 和VSMCs細胞,按照細胞計數1∶1比例進行混合培養,次日進行實驗。
2動物分組和方法
2.1失血性休克后eNOS的蛋白表達變化 將48只大鼠麻醉后右側股動脈插管接血壓計[6],隨機分組為:正常對照組、休克10 min、30 min、1 h、2 h、4 h組,每組8只,制作40 mmHg失血性休克模型,并按各組要求維持不同休克時間。休克完成后,活殺大鼠取SMA組織,常規Western blotting技術測定eNOS的蛋白表達。Quantity One軟件分析蛋白條帶,以eNOS/β-actin吸光度值之比反映eNOS蛋白表達。
2.2eNOS抑制劑對Ang-1和Ang-2調節缺氧早期和晚期血管反應性的影響 取大鼠104只,麻醉后活殺取SMA一級分支血管環,隨機分為:正常對照組、缺氧10 min組、Ang-1+缺氧10 min組、Ang-2+缺氧10 min組、eNOS抑制劑+缺氧10 min組、eNOS抑制劑+Ang-1+缺氧10 min組、eNOS抑制劑+Ang-2+缺氧10 min組、缺氧4 h組、Ang-1+缺氧4 h組、Ang-2+缺氧4 h組和eNOS抑制劑+缺氧4 h組、eNOS抑制劑+Ang-1+缺氧4 h組、eNOS抑制劑+Ang-2+缺氧4 h組,每組8只血管環。將血管環浸入無糖Krebs-Henseleit(K-H)液中,進行藥物處理,采用Ang-1和Ang-2的濃度均為200 μg/L,eNOS抑制劑L-N5-亞氨基乙基鳥氨酸二鹽酸鹽(L-N5-iminoethyl ornithine dihydrochloride,L-NIO)濃度為1×10-4mol/L。然后將血管環置于缺氧罐中,依次充入缺氧氣體(5%CO2和95%N2)15 min和夾閉10 min,循環5次,最后1次充氣完成后,按各實驗組要求分別夾閉10 min或4 h完成缺氧[6]。再取出缺氧處理完成的血管環,將其固定在微血管肌動描記儀上,浸入K-H液中,采用累積濃度法檢測微血管環對梯度濃度去甲腎上腺素(norepinephrine,NE)的反應性,作量-效曲線,曲線擬合法求NE的最大收縮力 (Emax),以NE的Emax評價血管反應性[7]。
2.3Ang-1和Ang-2調節缺氧早期血管高反應性時eNOS的蛋白表達變化和NO的含量變化 取混合培養的VECs 和VSMCs細胞32瓶,隨機分為:正常對照組、缺氧10 min組、Ang-2+缺氧10 min組、Tie-2抑制劑+缺氧10 min組、Akt抑制劑+缺氧10 min組、p38MAPK抑制劑+缺氧10 min組、ERK抑制劑+缺氧10 min組和eNOS抑制劑+缺氧10 min組,每組4瓶。先將細胞培養基換做無血清培養基,進行藥物處理,Ang-1和Ang-2的濃度均為200 μg/L,Tie-2抑制劑為1∶100封閉抗體,Akt抑制劑 1×10-5mol/L,p38 MAPK抑制劑SB203580 1×10-5mol/L,ERK抑制劑PD98059 1×10-5mol/L,eNOS抑制劑L-NIO 1×10-4mol/L。再進行不同時間缺氧處理(方法同前面SMA的缺氧處理)。完畢后取細胞培養上清,采用一氧化氮檢測試劑盒測定NO含量,收集細胞用于測定eNOS的蛋白表達。
3統計學處理
結 果
1失血性休克后eNOS的蛋白表達變化
eNOS在正常時表達很低(與β-actin的比值為0.0720),失血性休克后表達逐漸增高。休克1 min、30 min、1 h、2 h和4 h時分別增高至0.1407、0.1509、0.2173、0.2462和0.2571 (P<0.01),見圖1。
2eNOS抑制劑對Ang-1和Ang-2調節缺氧早期和晚期血管反應性的影響
eNOS抑制劑可顯著抑制缺氧10 min的血管高反應性,表現為量效曲線右移,NE的Emax由13.479 mN降低至9.043 mN (P<0.01),也可以顯著抑制Ang-1對缺氧10 min血管高反應性的維持作用,表現為量效曲線右移,NE的Emax由15.283 mN降低至11.219 mN (P<0.01),但不改變Ang-2降低缺氧10 min血管高反應性作用,也不改變缺氧4 h的血管低反應性和Ang-1、Ang-2對缺氧4 h血管反應性的調節作用,見圖2。


圖1失血性休克后eNOS蛋白表達
3Ang-1和Ang-2調節缺氧早期血管反應性時eNOS的蛋白表達變化和NO含量變化
缺氧10 min的eNOS表達較正常對照顯著增高(P<0.01),Ang-2、Tie-2抑制劑和Akt抑制劑可抑制其增高,使其由缺氧10 min的0.1354分別降低至0.0848、0.0724和0.0993 (P<0.01),p38 MAPK和ERK抑制劑對缺氧10 min的eNOS表達無顯著影響,見圖3。
NO含量在缺氧10 min即顯著增高,Ang-2以及Tie-2、Akt和eNOS抑制劑可抑制其增高,使其分別降低41.88%、68.91%、70.69%和67.42% (P<0.01),p38 MAPK和ERK抑制劑對其無顯著影響,見圖4。
討 論
失血性休克后血管反應性呈早期增高和晚期降低的雙相變化[8],其發生機制尚不清楚。本實驗室前期研究發現,血管生成素家族中的Ang-1和Ang-2在失血性休克后的SMA中呈時間上的差異表達(即Ang-1蛋白表達在休克早期增高,晚期降低;Ang-2蛋白表達在休克早期變化不大,在晚期逐漸增高),并通過Tie-2受體參與了失血性休克血管反應性雙相變化的形成[1],Akt、p38 MAPK和ERK幾種分子參與了其調節過程,但具體調節機制如何,尚無文獻報道。
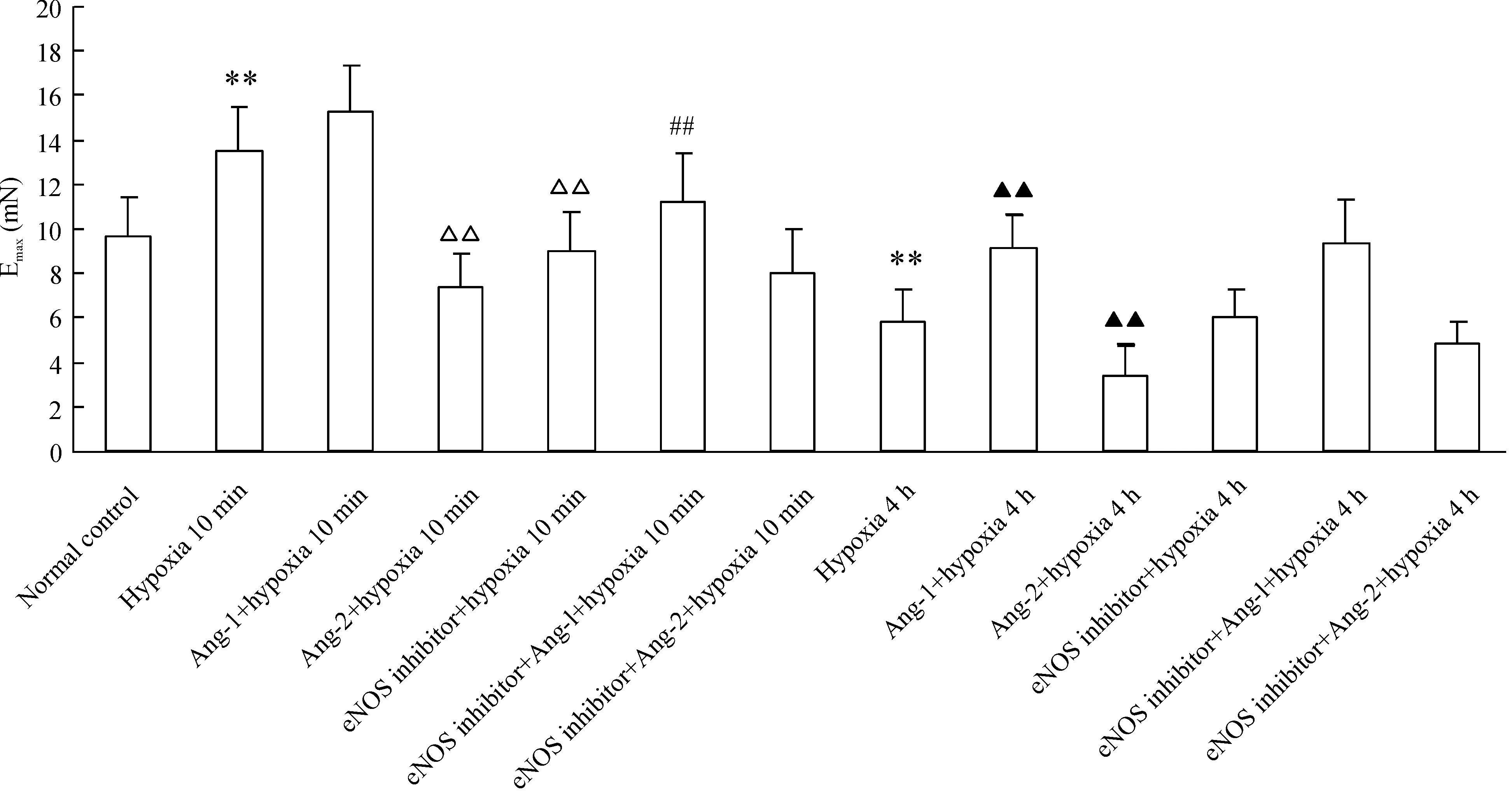
圖2eNOS抑制劑對Ang-1和Ang-2調節缺氧早期和晚期血管反應性的影響
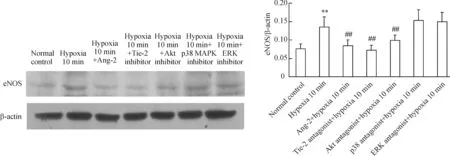
圖3Ang-1和Ang-2調節缺氧早期血管反應性時eNOS蛋白的表達
根據基礎研究,Ang-1和Ang-2激活VECs上的Tie-2受體之后,可經PI3K激活Akt,還可經過銜接蛋白Grb2活化p38 MAPK和ERK,再通過調節eNOS產生NO[2-3]。NO是目前已知的一種對血管舒縮反應性有重要調節作用的分子,少量NO可增高血管收縮反應性;過量NO可過度開放KATP和BKCa,導致VSMCs膜超極化,還可降低肌球蛋白輕鏈磷酸化,導致VSMCs鈣失敏,從而降低血管舒縮反應性[4]。且有研究發現,在LPS誘導內毒素休克的人臍靜脈內皮細胞,LPS通過磷酸化和活化Akt、p38 MAPK和ERK,激活eNOS,產生NO,調節內毒素休克的血管反應性[9-10]。那么,Ang-1和Ang-2是否通過Akt、p38 MAPK和ERK,調節eNOS產生NO,來調節失血性休克血管反應性的雙相變化?
本實驗研究發現,eNOS抑制劑可顯著抑制缺氧10 min的血管高反應性,也可以顯著抑制Ang-1對缺氧10 min血管高反應性的維持作用,但不改變缺氧4 h的血管低反應性和Ang-1、Ang-2對缺氧4 h血管反應性的調節作用,提示eNOS主要參與了Ang-1和Ang-2對失血性休克早期血管高反應性的調節。失血性休克后SMA中的eNOS蛋白表達逐漸增高;在缺氧培養的VECs和VSMCs混合細胞中,Ang-2以及Tie-2和Akt抑制劑可顯著降低缺氧10 min的eNOS蛋白表達增高和NO含量增高,p38 MAPK和ERK抑制劑對其沒有顯著影響,提示失血性休克早期,Ang-1和Ang-2通過Akt調節eNOS的蛋白表達和NO產生,來調節血管高反應性。

圖4Ang-1和Ang-2調節缺氧早期血管反應性時NO含量的變化
然而,本實驗還發現,盡管eNOS蛋白表達在休克晚期也是增高的,卻對于Ang-1和Ang-2調節休克晚期的血管低反應性作用沒有顯著影響,提示Akt-eNOS-NO信號途徑并不介導Ang-1和Ang-2對休克晚期血管低反應性的調節,那么Ang-1和Ang-2對休克晚期血管低反應性的調節又是通過什么分子和信號通路實現的,還需要進一步研究。
[1] 徐 競,李 濤,楊光明,等. Ang-1、Ang-2差異表達在大鼠失血性休克血管反應性雙相變化中的作用[J]. 中國病理生理雜志, 2010,26(9): 1684-1688.
[2] Brindle NP,Saharinen P,Alitalo K. Signaling and functions of angiopoietin-1 in vascular protection[J]. Circ Res, 2006,98 (8): 1014-1023.
[3] Chen JX, Lawrence ML, Cunningham G, et al. HSP90 and Akt modulate Ang-1 induced angiogenesis via NO in coronary artery endothelium[J]. J Appl Physiol, 2004,96(2): 612-620.
[4] Tokuno S, Chen F, Pernow J, et al. Effects of spontaneous or induced brain ischemia on vessel reactivity: the role of inducible nitric oxide synthase[J]. Life Sci, 2002,71(6): 679-692.
[5] Yang G, Xu J, Li T, et al. Role of V1areceptor in AVP -induced restoration of vascular hyporeactivity and its relationship to MLCP-MLC20phosphorylation pathway[J]. J Surg Res,2010,161(2):312-320.
[6] Li T, Liu L, Liu J, et al. Mechanism of Rho kinase regulation of vascular reactivity following hemorrhagic shock in rats[J]. Shock,2008,29 (1):65-70.
[7] Xu J, Yang G, Li T, et al. Involvement of CPI-17 and zipper-interacting protein kinase in the regulation of protein kinase C-α,protein kinase C-ε on vascular calcium sensitivity after hemorrhagic shock[J]. Shock, 2010, 33(1): 49-55.
[8] Liu LM, Dubick MA. Hemorrhagic shock-induced vascular hyporeactivity in the rat: relationship to gene expression of nitric oxide syntheas, endothelin-1, and select cytokines in corresponding organs[J]. J Surg Res,2005,125(2): 128-136.
[9] Connelly L, Madhani M, Hobbs AJ. Resistance to endotoxic shock in endothelial nitric oxide synthase (eNOS) knock-out mice: a proinflammatory role for eNOS-derived noinvivo[J]. J Biol Chem,2005,280(11): 10040-10046.
[10]Koide N, Mu MM, Hassan F, et al. Lipopolysaccharide enhances interferon-γ-induced nitric oxide (NO) production in murine vascular endothelial cells via augmentation of interferon regulatory factor-1 activation[J]. J Endotoxin Res, 2007,13(3): 167-175.
Akt-eNOS-NOsignalpathwaymediatesregulatoryeffectofAng-1andAng-2onvascularhyperreactivityinearlyhemorrhagicshockrats
XU Jing, LAN Dan, YANG Guang-ming, LI Tao, LIU Liang-ming
(StateKeyLaboratoryofTrauma,BurnandCombinedInjury,theSecondDepartmentofResearchInstituteofSurgery,DapingHospital,theThirdMilitaryMedicalUniversity,Chongqing400042,China.E-mail:Liuliangming2002@yahoo.com)
AIM: To observe the role of endothelial nitric oxide synthase (eNOS) in the regulatory effect of angiopoietin-1 (Ang-1) and angiopoietin-2 (Ang-2) on the biphasic change of vascular reactivity after hemorrhagic shock in rats.METHODSThe protein expression of eNOS was measured in the superior mesenteric artery (SMA) after hemorrhagic shock by Western blotting. The effect of eNOS inhibitor on the vascular reactivity of SMA treated with Ang-1 and Ang-2 in the early (hyperreactivity) and late (hyporeactivity) periods of hypoxia were observed via an isolated organ perfusion system. The protein levels of eNOS in the hypoxic mixture of vascular endothelial cells (VECs) and vascular smooth muscle cells (VSMCs), and the concentration of nitric oxide (NO) in the medium supernatant of the mixture cells treated with Ang-1, Ang-2 and the inhibitors of Tie-2, Akt, p38 MAPK and ERK were measured.RESULTSThe protein expression of eNOS in SMA was low in normal control group, and increased significantly after hemorrhagic shock, which was 1.84, 3.55, 4.75, 5.96 and 6.33 folds of the normal control level in shock 10 min, 30 min, 1 h, 2 h and 4 h groups, respectively (P<0.01). Inhibitor of eNOS decreased the vascular hyperreactivity in hypoxia 10 min group, in which the Emaxof norepinephrine (NE) was decreased from 13.479 mN to 9.043 mN (P<0.05). It also repressed the maintenance effect of Ang-1 on vascular reactivity in hypoxia 10 min group, in wihich the Emaxof NE was decreased from 15.283 mN to 11.219 mN (P<0.01). The effect of Ang-2 on the vascular hyperreactivity in hypoxia 10 min group, the vascular hyporeactivity in hypoxia 4 h group, or the effect of Ang-1 or Ang-2 on the vascular reactivity in hypoxia 4 h group did not change. The protein expression of eNOS was increased 10 min after hypoxia as compared with the normal control, which was decreased by Ang-2 and the inhibitors of Tie-2 and Akt (P<0.01), but was not decreased by p38 MAPK and ERK inhibitors. The concentration of NO in the medium supernatant was increased 10 min after hypoxia, and was significantly decreased by Ang-2 and the inhibitors of Tie-2, Akt and eNOS, while the inhibitors of p38 MAPK and ERK had no influence on it.CONCLUSIONAng-1 and Ang-2 regulate the vascular hyperreactivity in the early hemorrhagic shock rats through Akt-eNOS-NO pathway.
Hemorrhagic shock; Vascular reactivity; Biphasic change; Angiopoietin-1; Angiopoietin-2; Endothelial nitric oxide synthase
R605.971
A
10.3969/j.issn.1000-4718.2012.09.004
