三環已基錫2-氨基-6-氯苯甲酸酯配合物的合成、結構及量子化學研究
2012-11-13 05:51:04張復興王劍秋鄺代治馮泳蘭陳志敏許志鋒
無機化學學報 2012年9期
張復興 王劍秋 鄺代治 馮泳蘭 陳志敏 許志鋒
(衡陽師范學院化學與材料科學系,功能金屬有機材料湖南省普通高等學校重點實驗室,衡陽 421008)
三環已基錫2-氨基-6-氯苯甲酸酯配合物的合成、結構及量子化學研究
張復興*王劍秋 鄺代治 馮泳蘭 陳志敏 許志鋒
(衡陽師范學院化學與材料科學系,功能金屬有機材料湖南省普通高等學校重點實驗室,衡陽 421008)
三環己基氫氧化錫與2-氨基-6-氯苯甲酸按物質的量比1∶1在苯溶劑中反應合成了三環己基錫2-氨基-6-氯苯甲酸酯。經X-射線衍射方法測定了其晶體結構,配合物屬單斜晶系,空間群為P21/c,晶體學參數a=0.952 00(4)nm,b=0.918 55(3)nm,c= 2.909 12(12)nm,β=93.187(2)°,V=2.539 97(17)nm3,Z=4,Dc=1.409 g·cm-3,μ(Mo Kα)=11.31 cm-1,F(000)=1 112,R1=0.038 2,wR2= 0.098 6。中心錫與環己基碳原子和氧原子構成畸型四面體。對其結構進行量子化學從頭計算,探討了配合物的穩定性、分子軌道能量以及一些前沿分子軌道的組成特征。通過循環伏安法研究了其電化學性質。
三環己基錫2-氨基-6-氯苯甲酸酯;合成;晶體結構;從頭計算;循環伏安
有機錫羧酸酯由于具有結構的多變性、豐富的反應性、較強的生物活性和催化活性,多年來一直引起人們的興趣[1-5]。然而,由于有機錫化合物的高毒性,又使它們的應用受到了一定的限制。相關研究表明,有機錫化合物的生物活性與中心錫原子的構型有關,而中心錫原子的構型決定于直接與錫原子相連的烴基的結構和配體的類型[6-11]。功能化的羧酸配體能極大的改變錫原子的配位方式,顯著的影響有機錫羧酸酯的生物活性,從而調節其毒性與生物活性之間的平衡。當以芳酸為配體,在芳環上引入功能化基團時,它既可利用立體效應,也可利用其電子效應來極大地改變有機錫羧酸酯的物理與化學性能。近年來我們合成了一系列有機錫芳香羧酸酯化合物,并研究了它們的結構和性能[12-17]。為了更進一步揭示有機錫化合物結構與性能的關系,本文合成了三環己基錫2-氨基-6-氯苯甲酸酯,通過元素分析、紅外光譜和核磁共振氫譜進行了表征,用X-射線單晶衍射測定了晶體結構,對其結構進行量子化學從頭計算,探討了配合物的穩定性、分子軌道能量以及一些前沿分子軌道的組成特征。并利用循環伏安法對其進行了電化學性質分析。
1 實驗部分
1.1 試劑與儀器
日本島津 FTIR-8700紅外光譜儀(4 000~400 cm-1,KBr),PE-2400(Ⅲ)元素分析儀,Bruker SMART APEXⅡ單晶衍射儀,X4數字顯微熔點測定儀。所用試劑均為分析純。
1.2 實驗過程
在50mL圓底燒瓶中,加入0.770 g(2mmol)三環己基氫氧化錫、0.341 g(2mmol)2-氨基-6-氯苯甲酸、40 mL苯,在電磁攪拌下加熱回流分水反應5 h。趁熱過濾除去不溶性固體,濾液旋轉蒸發除去部分溶劑,放置析出棕色固體,用適當的溶劑重結晶得棕色透明晶體0.748 g,產率69.49%。熔點:104~106℃。紅外光譜主要吸收峰:3 377.1(m),3 055.5 (m),2920.0(s),11614.3(s),1577.7(m),1468.5(m),1444.6(m),1357.8(s),589.0(w),453.2(w)cm-1。1H NMR (CDCl3,400 MHz),δH:1.31~2.00(m,3×11H,Cp-H),4.97(s,2H,Ar-NH2),6.55(d,1H,3-Ar-H),6.73(d,1H, 5-Ar-H),7.01(t,1H,4-Ar-H)。元素分析(C25H38ClNO2Sn),計算值(%):C,55.74;H,7.06;N,2.60。實測值(%):C,56.08;H,7.12;N,2.54。
1.3 晶體結構測定
選取尺寸為0.2 mm×0.2 mm×0.2 mm單晶體,在Bruker SMART APEXⅡ單晶衍射儀上進行衍射實驗,在296(2)K下,用石墨單色化的Mo Kα(λ= 0.071 073 nm)射線,以ω-2θ方式掃描收集數據。在2.14°≤θ≤27.61°范圍內共收集18 172個衍射點,其中獨立衍射點4 473個(Rint=0.032 2),可觀察衍射點4 126個(I>2σ(I))。全部數據經Lp校正和吸收校正,以直接法進行晶體結構解析。部分非氫原子坐標隨后用差值Fourier合成法確定,理論加氫計算法給出氫原子位置坐標。用SHELX97程序以全矩陣最小二乘法對非氫原子坐標及其各向異性熱參數進行修正,殘差因子R1=0.038 2,wR2=0.098 6。
CCDC:888049。
2 結果與討論
2.1 晶體結構描述
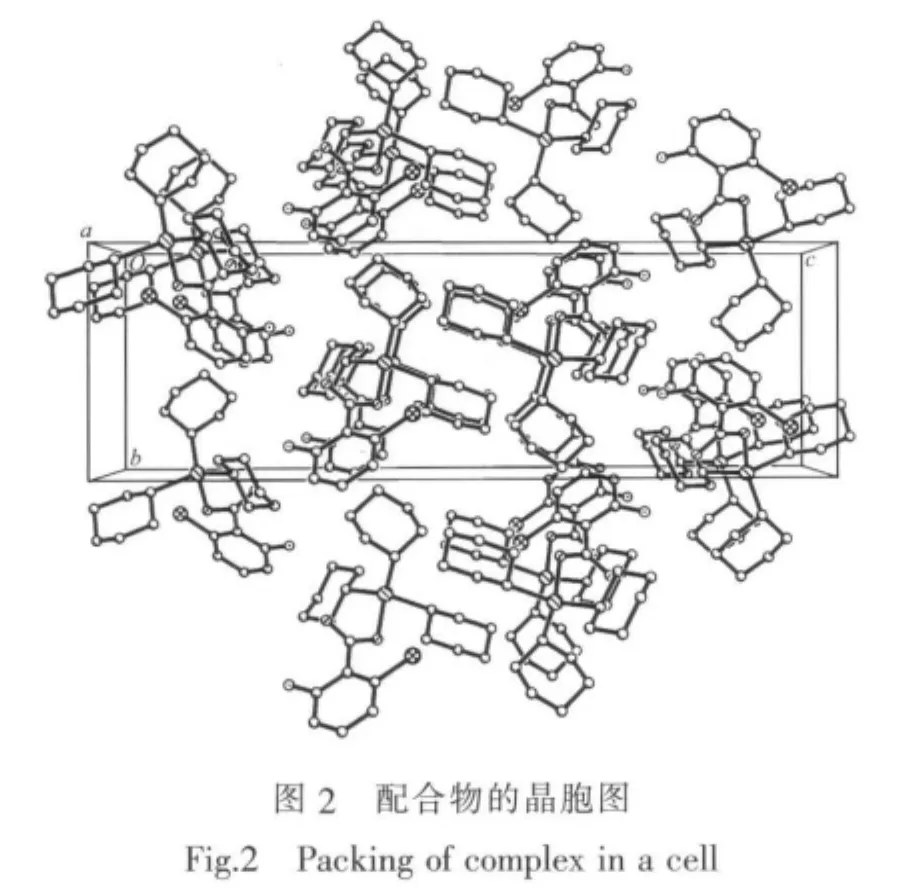
配合物屬單斜晶系,空間群為P21/c,晶體學參數a=0.952 00(4)nm,b=0.918 55(3)nm,c=2.909 12(12) nm,β=93.187(2)°,V=2.539 97(17)nm3,Z=4,Dc=1.409 g·cm-3,μ(Mo Kα)=11.31cm-1,F(000)=1112,R1=0.038 2, wR2=0.0986;Δρmax=743 e·nm-3,Δρmin=-493 e·nm-3。配合物的主要鍵長和鍵角列于表1,分子結構見圖1,晶胞中分子堆積見圖2。
從圖1和結構參數可知,中心錫原子與3個環己碳原子及1個羧基氧原子相連形成四面體構型。由于與2-氨基-6-氯苯甲酰基的空間效應引起了3個環己基處于不同的空間環境,使得環上C-C鍵在0.142 9~0.158 3 nm之間形成不對稱六元環,且四面體的鍵角均偏離了正四面體角。3個 Sn-C鍵鍵長分別為Sn(1)-C(1)0.215 4(3)nm、Sn(1)-C(7) 0.215 4(43)nm和Sn(1)-C(3)為0.215 6(3)nm,較為接近,與中心錫原子間的鍵角分別為C(1)-Sn(1)-C(7) 110.24(14)°、C(1)-Sn(1)-C(13)為 112.69(11)°、C(7)-Sn(1)-C(13)123.54(14)°,都比正四面體鍵角大;Sn-O鍵鍵長為0.207 49(19)nm,與Sn(1)-C鍵的鍵角分別為C(1)-Sn(1)-O(1)為94.78(9)°、C(7)-Sn(1)-O(1)為102.56(13)°、C(13)-Sn(1)-O(1)108.48(9)°,都比正四面體角小;這種鍵角的變化是由于環己基的空間位阻所致。這種空間排列就決定了中心錫與環己基碳原子和氧原子構成畸型四面體。

表1 配合物的主要鍵長和鍵角Table 1 Selected bond distances(nm)and selected bond angles(°)of the title com plex

2.2 配合物的能量和前沿分子軌道組成
根據晶體結構的原子坐標,運用Gaussian 98W程序和HF/lanl2dz基組水平,計算得到分子的總能量為-1 190.151 550 3 a.u,最高占據軌道能量為-0.300 64 a.u,最低空軌道能量為0.102 67 a.u。可見總能量和最高占有軌道能量均較低,最高占據軌道與最低未占軌道的能量間隙ΔE=0.4.331 a.u,表明分子結構穩定。從氧化還原轉移的角度分析,HOMO能級較低,難以給出電子而被氧化。
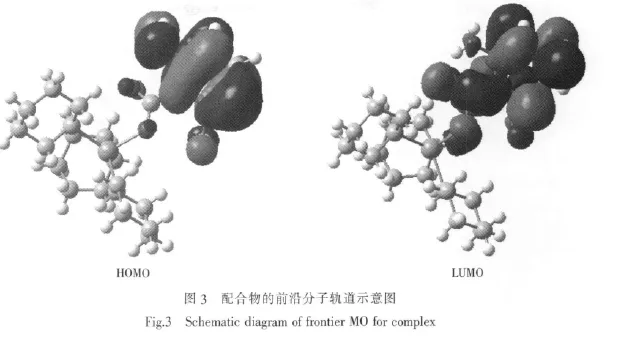
為探索化合物的電子結構與成鍵特征,對化合物分子軌道進行分析,用參與組合的各類原子軌道系數的平方和來表示該部分在分子軌道中的貢獻,并經歸一化。把化合物原子分為七部分:(a)錫原子Sn;(b)羧基氧原子O;(c)羧基碳原子C(Ⅲ);(d)苯環碳原子C(Ⅲ);(e)環己環碳原子C(Ⅲ); (f)氯原子Cl;(g)氮原子N;(h)氫原子H。前沿占有軌道和未占有軌道各取5個,計算結果如表2和圖3所
表2和圖3顯示分子的成鍵特征:①前沿占有分子軌道中,苯環碳對分子軌道的貢最大,為67.3%,說明苯環具有高度的共軛離域性和穩定性;其次是氨基氮原子為28.24%,說明氨基氮原子與苯環具有良好的共軛性。② 前沿占有分子軌道中,氯原子對分子軌道的貢獻僅為2.73%,比氮原子小得多,說明氯原子能與苯環的共軛程度比氮原子小,與原子軌道重疊能量相近的原則相一致。因氮原子是2p軌道與苯的大π鍵共軛,而氯原子是3p與苯的大π鍵共軛,兩者的能量相差較大。③前沿占有分子軌道中,羧基氧和羧基碳對分子軌道的貢獻都較小,分別為1.07%和0.29%,說明羧基與苯環的共軛程度小,這主要是在羧基的2個鄰位有空間體積較大的氨基和氯原子,由于空間位阻使羧基難以與苯環共平面。④比較HOMO與LUMO的各類原子軌道成份,可以看出,當電子從HOMO激發到LUMO軌道時,主要是氮原子和氯原子能上的電子通過苯環向羧基和錫原子轉移,苯環既是電子轉移的橋梁也是電子轉移的受體。
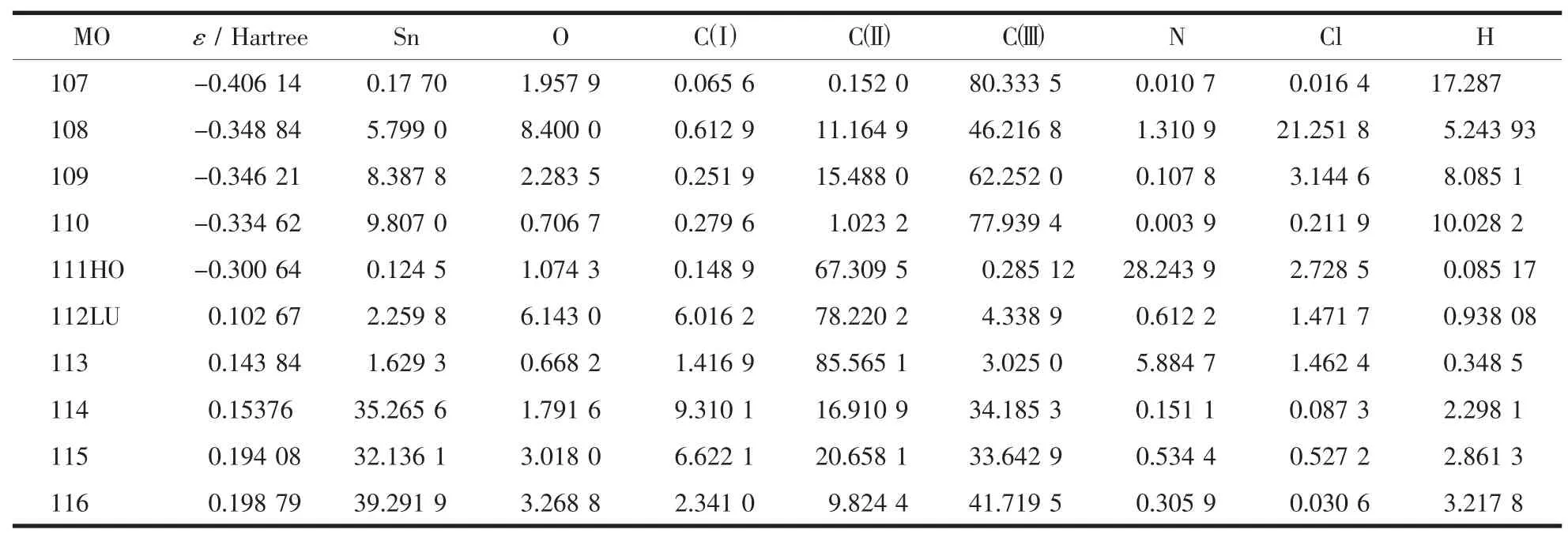
表2 分子軌道組成Table 2 Calculated some frontier molecular orbitals composition(%)of com plex
2.3 電子結構研究
由Mulliken布居分析得到結構單元的原子電荷如表3所示,顯示出電荷布居的一些規律和特征:①中心錫原子失去較多電子而荷1.766 397的正電,所有氫原子均荷正電。②2個氧原子中O(1)、O(2)荷0.751 070和0.506 465負電,這是因為O(1)與Sn(1)形成了Sn-O鍵,從錫原子上獲得了電子,而O(2)沒有與Sn(1)成鍵,因此,羧基與錫原子是單齒配位,與X-射線衍射的結論相一致。③碳原子中除羧基碳、與羧基相連的苯環碳及與氨基相連的苯環碳荷正電外,其它碳原子均荷負電,并且符合電荷分布一般規律。

表3 配合物的部分原子電荷Table 3 Atom ic charge populations of com pound
2.3 配合物的循環伏安(CV)
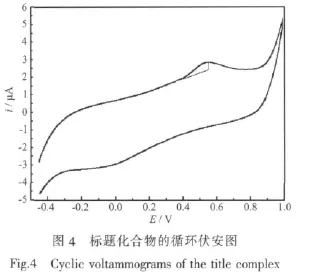
為考察所合成配合物在溶液中的電化學性能,采用常規三電極體系:玻碳電極為工作電極,鉑電極為輔助電極,SCE為參比電極,以乙醇為溶劑,配合物的濃度為 1.0μmol·L-1,于室溫下進行電化學性質測定。在-0.500~1.000 V范圍內,以100mV· s-1速度進行循環伏安掃描,所得結果如圖4。從圖可看出,配合物無還原峰,僅有1個氧化峰,其峰電位為0.536 V,由此說明配合物在電極上的電子轉移是不可逆的。
[1]Chandrasekhar V,Thirumoorthi R,Metre R K,et al.J. Organomet.Chem.,2011,696:600-606
[2]Effendy,Marchetti F,Marinelli A,et al.J.Inorg.Chem. Acta.,2011,366:388-393
[3]Hanif M,Hussain M,Ali S,et al.J.Polyhedron,2010,29: 613-619
[4]ZHANG Xiao-Yan(張曉燕),YANG Guang(楊光),ZHANG Jun(張俊),et al.Chem.J.Chinses Universities(Gaodeng Xuexiao Huaxue Xuebao),2010,31(6):1162-1166
[5]Siddiqi ZA,Shahid M,Kumar S,etal.J.Organomet.Chem., 2009,694:3768-3774
[6]Ruan B F,Tian Y U,Zhou H P,et al.J.Chim.Acta,2011, 365:302-308
[7]ZHANGFu-Xing(張復興),KUANGDai-Zhi(鄺代治),WANG Jian-Qiu(王劍秋),etal.Chinese J.Inorg.Chem.(WujiHuaxue Xuebao),2006,22(7):1321-1326
[8]YINHan-Dong(尹漢東),WANGChuan-Hua(王傳華),WANG Yong(王勇),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2002,18(2):201-204
[9]YAN Wen-Hua(閆文華),KANGWan-Li(康萬利),LI Jin-Huan(李金環).Chinese J.Appl.Chem.(Yingyong Huaxue), 2007,24(6):660-664
[10]Shujha S,Shah A,Rehman Z U,et al.J.Eur.J.Med. Chem.,2010,45:2902-2911
[11]ZHANGFu-Xing(張復興),KUANGDai-Zhi(鄺代治),WANG Jian-Qiu(王劍秋),etal.Chinese J.Org.Chem.(YoujiHuaxue), 2008,28(8):1457-1461
[12]ZHANG Fu-Xing(張復興),WANG Jian-Qiu(王劍秋),KUANG Dai-Zhi(鄺代治),etal.Chinese J.Inorg.Chem.(WujiHuaxue Xuebao),2011,27(8):1591-1595
[13]ZHANGFu-Xing(張復興),WANG Jian-Qiu(王劍秋),KUANG Dai-Zhi(鄺代治),etal.Chinese J.Inorg.Chem.(WujiHuaxue Xuebao),2011,27(6):1111-1115
[14]YU Jiang-Xi(庾江喜),KUANG Dai-Zhi(鄺代治),YIN Du-Lin(尹篤林),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2010,26(8):1507-1510
[15]ZHANG Fu-Xing(張復興),WANG Jian-Qiu(王劍秋),KUANG Dai-Zhi(鄺代治),etal.Chinese J.Inorg.Chem.(WujiHuaxue Xuebao),2009,25(2):213-217
[16]ZHANG Fu-Xing(張復興),WANG Jian-Qiu(王劍秋),KUANG Dai-Zhi(鄺代治),et al.Chinese.J.Appl.Chem.(Yiyiong Huaxue),2009,26(6):662-666
[17]Zhang F X,Wang JQ,Kuang D Z,et al.Chinese J.Struct. Chem.,2010,29(10):1529-1535
Synthesis,Crystal Structure and Quantum Chem istry of the Tricyclohexyltin 2-Am ino-6-chloro Benzoate
ZHANG Fu-Xing*WANG Jian-Qiu KUANG Dai-Zhi FENG Yong-Lan CHEN Zhi-Ming XU Zhi-Feng
(Departmentof Chemistry and Material Science,Hengyang Normal University;Key Laboratory of Functional Organometallic Materials of Hengyang Normal University,College of Hunan Province,Hengyang,Hunan 421008,China)
The tricyclohexyltin 2-amino-6-chlorobenzoate was synthesized by the reaction of the tricyclohexyltin hydroxide with the 2-amino-6-chlorobenzoic acid.Its structure has been determined by X-ray single crystal diffraction.The crystal belongs tomonoclinic with space group P21/c,a=0.952 00(4)nm,b=0.918 55(3)nm,c= 2.909 12(12)nm,β=93.187(2)°,V=2.539 97(17)nm3,Z=4,Dc=1.409 g·cm-3,μ(Mo Kα)=11.31 cm-1,F(000)=1 112, R1=0.038 2,wR2=0.098 6.The tin atom has a distorted tetrahedral geometry.The study on title complex has been performed,with quantum chemistry calculation by means of G98W package and taking Lanl2dz basis set.The stabilities of the complex,the orbital energies and composition characteristics of some frontiermolecular orbitals have been investigated.The electrochemistry propertieswere studied by cyclic voltammetry.CCDC:888049.
tricyclohexyltin 2-amino-6-chlorobenzoate;synthesis;crystal structure;ab initiomethod;Cyclic voltammetry
O612.43+2
A
1001-4861(2012)09-1890-05
2012-01-29。收修改稿日期:2012-06-06。
湖南省教育廳重點項目(No.10A014、10K010)、湖南省自然科學基金項目(No.11JJ3021)、湖南省重點學科基金資助項目。
*通訊聯系人。E-mail:zfx8056@yahoo.com.cn;會員登記號:S060018907M。
