UPLC法與HPLC法測定加替沙星微球有關物質及微球的光穩定性評價Δ
2012-11-21 07:33:08林華慶張肖玲廣東藥學院藥物研究所廣東省藥物新劑型重點實驗室廣州510006
中國藥房 2012年33期
鄧 紅,張 蜀,林華慶,張肖玲,田 璐(廣東藥學院藥物研究所/廣東省藥物新劑型重點實驗室,廣州510006)
UPLC法與HPLC法測定加替沙星微球有關物質及微球的光穩定性評價Δ
鄧 紅*,張 蜀#,林華慶,張肖玲,田 璐(廣東藥學院藥物研究所/廣東省藥物新劑型重點實驗室,廣州510006)
目的:采用超高效液相色譜法(UPLC)與高效液相色譜法(HPLC)測定加替沙星微球的有關物質,并進行微球的光穩定性評價。方法:UPLC法、HPLC法的色譜柱分別為Waters ACQUITY uplc?HSS T3、Diamosil C18(2),流速分別為0.6、1.0mL·min-1,進樣量分別為5、50μL,流動相均為三乙胺磷酸溶液(pH 4.3)-乙腈(88∶12),檢測波長均為293nm;另取微球和原料藥在溫度為25℃、濕度為60%、光照強度為6600lx下放置5、10d后,采用UPLC法考察雜質譜變化情況。結果:UPLC法和HPLC法檢測加替沙星的最低檢測限分別為0.001、4.1ng,最低定量限分別為0.002、15.3ng,檢測時間分別為8、40min左右;微球和原料藥經光照后雜質譜相似,但原料藥中的部分雜質含量明顯增加。結論:在擬定的色譜條件下,與HPLC法比較,UPLC法更快速、靈敏度更高;加替沙星制成微球后對光的穩定性增加。
超高效液相色譜法;高效液相色譜法;加替沙星微球;有關物質;光穩定性
加替沙星為第4代喹諾酮類抗菌藥,其抗菌譜廣,對G+細菌、衣原體、支原體等都有強大的殺菌作用,且光毒性較弱,對上述病原體引起的各種感染治療均有效[1]。本課題研制的加替沙星微球將進一步制成關節腔埋植劑,用于治療骨髓炎。迄今為止,加替沙星及其制劑的質量標準均未在各國藥典收載,在藥品質量標準中,對有關物質的控制是質量控制的重要環節,是穩定性考察的重要指標,同時也是藥品評價的重要參數。超高效液相色譜(UPLC)是近年來發展迅速的基于小顆粒填料的液相色譜技術,相對于高效液相色譜(HPLC)法,能顯著改善色譜峰的分離度和檢測靈敏度,同時大大縮短分析周期[2,3],因此適用于微量復雜混合物如有關物質的分析。本文比較采用UPLC與HPLC法檢測加替沙星微球的有關物質,并比較加替沙星原料藥和微球對光的穩定性,為其埋植劑的開發提供參考,同時為加替沙星有關物質提供新的檢測方法。
1 儀器與試藥
UPLC H-Class液相色譜儀(美國Waters公司);Ultimate 3000液相色譜儀(美國Dionex公司)。
加替沙星原料藥及對照品(上虞京新藥業有限公司,批號:0703301,純度:99.5%);加替沙星微球(自制,批號:20110901、20110902、20110903,含量分別為:4.48%、4.32%、4.60%);乙腈為色譜純,水為重蒸餾水,三乙胺和磷酸均為分析純。
2 方法與結果
2.1 加替沙星微球的制備
按處方量稱取加替沙星和聚乳酸-羥基乙酸共聚物(PLGA,50∶50)(1∶12)置錐瓶中,加乙腈溶解得藥液。另取含0.25%司盤80(W/V)的液狀石蠟置具塞錐瓶中,800r·min-1攪拌下緩慢加入藥液,密塞;攪拌10min后400r·min-1繼續攪拌1h,緩慢滴入正己烷;3h后停止攪拌,用少量正己烷洗滌3次,干燥,即得。
2.2 色譜條件
HPLC和UPLC法均以三乙胺磷酸溶液(三乙胺溶液(1→100),用稀磷酸調pH(4.3±0.05))-乙腈(88∶12)為流動相,檢測波長為293nm,柱溫為室溫;HPLC法色譜柱采用Diamonsil-C18(2)(250mm×4.6mm,5μm),流速為1.0mL·min-1,進樣量為50μL;UPLC法色譜柱采用Waters ACQUITY uplc?HSS T3(50mm×2.1mm,1.8μm),流速為0.6mL·min-1,進樣量為5μL。
2.3 溶液的制備
2.3.1 微球供試品溶液。取加替沙星微球適量(約相當于加替沙星10mg),研細,精密稱定,置于50mL量瓶中,加乙腈6mL,超聲振搖使溶解,再加三乙胺磷酸溶液稀釋至刻度,搖勻,濾過,取續濾液,即得。
2.3.2 微球自身對照溶液。取上述供試品溶液1mL,置于100mL量瓶中,加流動相稀釋至刻度,即得。
2.3.3 原料藥供試品溶液。取加替沙星原料藥約10mg,精密稱定,置于50mL量瓶中,加流動相溶解并稀釋至刻度,即得。2.3.4陰性對照溶液。按處方工藝制備空白微球,精密稱取適量,依照“2.3.1”項下方法制備即得。
2.4 專屬性試驗
2.4.1 輔料干擾試驗。取微球供試品溶液、原料藥供試品溶液和陰性對照溶液各50μL注入HPLC儀,各5μL注入UPLC儀,記錄色譜圖,結果空白輔料不干擾加替沙星及其雜質的測定。HPLC和UPLC法檢測時間分別為40、8min左右,見圖1。
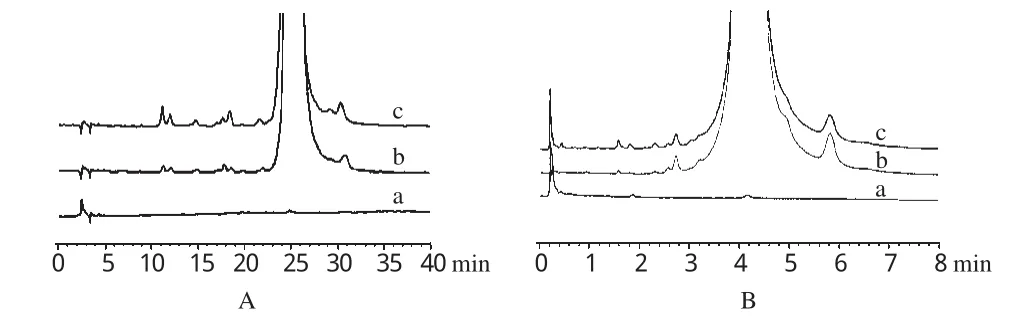
圖1 干擾試驗色譜圖A.HPLC法;B.UPLC法;a.陰性對照溶液;b.原料藥供試品溶液;c.微球供試品溶液Fig 1 HPLC and UPLC chromatograms of interfering test A.HPLC method;B.UPLC method;a.negative control solution;b.test sample solution of raw material;c.test sample solution of microspheres
2.4.2 強制降解試驗。取微球供試品溶液、原料藥供試品溶液和陰性對照溶液進行光照(紫外燈照射約48h)、高溫(沸水浴加熱2h,加乙腈補足減失體積)降解試驗;取微球、原料藥(約相當于加替沙星10mg)和空白微球適量,各3份,置50mL量瓶中,進行酸(加1mol·L-1鹽酸溶液1mL)、堿(加3mol·L-1NaOH溶液1mL)、氧化(加3%H2O2溶液1mL)降解試驗,再分別加乙腈6mL,超聲振搖使溶解,48h后加三乙胺磷酸溶液稀釋至刻度。取以上各溶液,濾過,取續濾液,采用2種方法進行分析。結果空白微球對上述5種破壞方法均較穩定;原料藥和微球對酸、堿破壞較穩定,光照破壞下檢出較多雜質。二者雜質譜相似,UPLC法對雜質的檢測能力較強,微球降解試驗的色譜圖詳見圖2(其他圖略)。
2.5 最低檢測限和最低定量限試驗
2.5.1 HPLC法。取原料藥,用流動相稀釋制成濃度分別為0.08176、0.3066μg·mL-1的溶液,進樣分析,記錄色譜圖。以信/噪為3計算最低檢測限為4.1ng(0.04%),以信/噪為10計算最低定量限為15.3ng(0.15%)。
2.5.2 UPLC法。取原料藥,用流動相稀釋制成濃度分別為0.2044、0.3066ng·mL-1的溶液,進樣分析,記錄色譜圖。以信/噪為3計算最低檢測限為0.001ng(0.0001%),以信/噪為10計算最低定量限為0.002ng(0.0002%)。
2.6 供試品溶液的穩定性考察
分別取原料藥、微球的供試品溶液注入UPLC儀,于0、1、2、6、8h測定,記錄峰面積,結果原料藥和微球中雜質1、2、6峰面積在2h后均增加,因此溶液制備后應在2h內測定。
2.7 樣品測定結果比較
取微球3批,依“2.3”項下方法制備成供試品溶液和自身對照溶液,按上述HPLC法和UPLC法測定。供試品溶液如顯雜質峰,單個雜質峰面積不得超過自身對照溶液的主峰面積的0.5倍(0.5%),各雜質峰面積的和不得大于自身對照溶液主峰面積(1.0%),測定結果見表1。
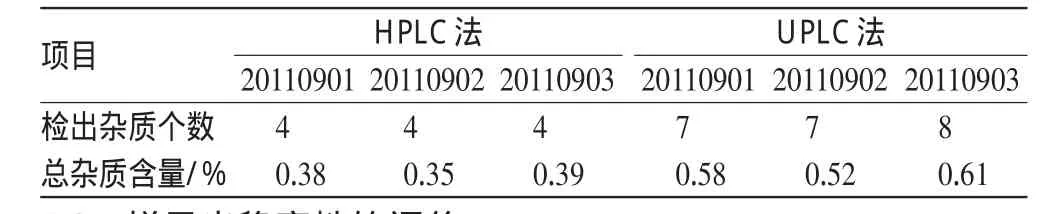
表1 2種方法檢測樣品中有關物質的結果比較Tab 1 Comparison of determination results of related substances by 2methods
2.8 樣品光穩定性的評價
取微球、原料藥和空白微球(陰性對照)適量,置于潔凈玻璃平皿中,放入智能型人工氣候箱,設置溫度為25℃、濕度為60%、光照強度為6600lx,于第5、10天時取樣,按UPLC方法進行雜質譜分析,比較微球和原料藥在光照射后的雜質譜變化,結果見圖3。
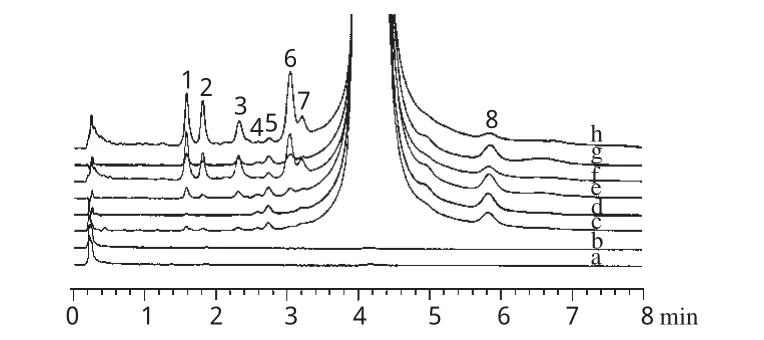
圖3 加替沙星原料藥/微球光降解試驗雜質色譜圖a.未光照陰性對照;b.光照10d陰性對照;c.未光照微球;d.未光照原料藥;e.光照5d微球;f.光照5d原料藥;g.光照10d微球;h.光照10d原料藥;1~8.未知雜質Fig 3 Impurity chromatograms of gatifloxacin raw material and microspheres in light degradation testa.untreated negative control;b.negative control treated by light for 10d;c.untreated microspheres;d.untreated raw material;e.microspherestreated by light for 5d;f.raw material treated by light for 5d;g.microspheres treated by light for 10d;h.raw material treated by light for 10d;1~8.unknown impurities
由圖3可見,微球和原料藥經光照試驗后,雜質譜相似,其中雜質4、5、8的峰面積基本沒有改變,原料藥的雜質1、2、3、6峰面積明顯增加,而微球相對較穩定。據此,可認為將加替沙星制成微球后,對光的穩定性提高,雜質1、2、3、6是與加替沙星光照穩定性密切相關的主要雜質。為此以雜質1、2、3、6和總雜質含量為指標,進一步比較加替沙星原料藥/微球光降解前后的雜質含量變化,結果表明微球處方工藝合理,詳見圖4。
3 討論
(1)檢測波長的確定。加替沙星在293nm波長有最大吸收[4],本試驗HPLC法和UPLC法均與二極管陣列檢測器(DAD)聯用,通過全波長掃描,在293nm波長處主峰和雜質峰的紫外吸收均較強,說明可檢出雜質的最大吸收也在293nm附近,故有關物質的檢測波長定在293nm。
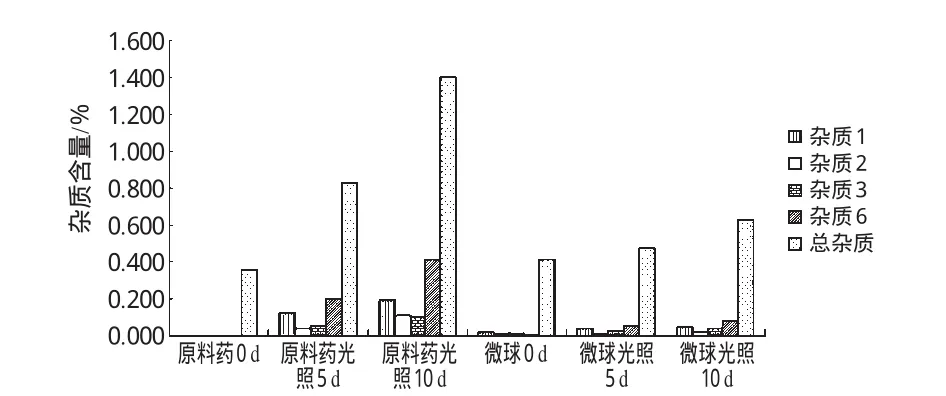
圖4 加替沙星原料藥/微球光照前后雜質含量變化比較Fig 4 Comparison of the contents of impurity in gatifloxacin raw material and microspheres before and after light
(2)溶液制備方法的選擇。本品主要以聚乳酸為輔料制成微球,由于加替沙星和聚乳酸均溶于乙腈,因此先用乙腈溶解微球,保證主藥完全釋出;考慮到與流動相的一致性,再按流動相比例加入pH為4.3的三乙胺溶液(1→100)。
(3)大多數喹諾酮藥物對酸、堿的穩定性較好,對光照穩定性差異較大。喹諾酮藥物的光穩定性不僅與藥物結構有關,且受處方工藝的影響。因此,對自制微球和原料藥光照穩定性進行了比較,結果微球和原料藥的雜質譜相似,說明雜質主要來自原料藥。但相對于原料藥,加替沙星制成微球后,對光的穩定性提高。
(4)本試驗比較了2種方法測定加替沙星微球的有關物質,與HPLC法相比,UPLC法具有檢測靈敏度更高、分析更快速、對雜質的檢測能力更強等優勢,在保證雜質檢出能力的前提下,分析效率更高,分析時間更短,溶劑損耗更低,降低了分析成本。因此,UPLC法特別適合藥物微量、復雜體系如有關物質的分析,值得推廣應用。
[1] 杜 唯,劉護魚.新一代氟喹諾酮類藥物——加替沙星[J].西北藥學雜志,2004,8(19):189.
[2] 郝桂明,唐素芳.超高效液相色譜在藥物分析中的應用[J].天津藥學,2009,21(6):64.
[3] 孫全樂,蔡廣知,貢濟宇.超高效液相色譜法測定人參中人參皂苷Re的含量[J].中國藥房,2012,23(3):258.
[4] 鄧 紅,梁健健,張 蜀,等.HPLC法測定加替沙星聚乳酸納米粒的含量[J].廣東藥學院學報,2008,24(3):236.
Evaluation of Light Stability of Gatifloxacin Microsphere and Related Substances by UPLC and HPLC
DENG Hong,ZHANG Shu,LIN Hua-qing,ZHANG Xiao-ling,TIAN Lu(Institute of Materia Medica of
Guangdong Pharmaceutical University/Guangdong Provincial Key Laboratory of Advanced Drug Delivery,Guangzhou 510006,China)
OBJECTIVE:To detect the related substances of Gatifloxacin microsphere by UPLC and HPLC,and to evaluate the light stability of microspheres.METHODS:UPLC was performed on Waters ACQUITY uplc?HSS T3column with the flow rate of 0.6mL·min-1and the injection volume was 5μL;HPLC was performed on Diamosil C18(2)column with the flow rate of 1.0mL·min-1and the injection volume was 50μL.The mobile phase was triethylamine-phosphoric acid solution(pH4.3)-acetonitrile(88∶12),and detection wavelength was set at 293nm.The chromatograms change of impurity was detected by UPLC after Gatifloxacin microsphere and raw material were kept at 25℃with moisture of 60%and light intensity of 6600lx for 5days and 10days.RESULTS:LOQ of gatifloxacin were 0.001ng and 4.1ng,and LOD were 0.002ng and 15.3ng for UPLC and HPLC.The determination duration were 8and 40min.The impurity chromatograms of microspheres and raw material were similar and the contents of some impurity in raw material increased.CONCLUSIONS:Under fixed chromatogram condition,UPLC method is proved to be more rapid and sensitive than HPLC method.The light stability of gatifloxacin is improved after being made into microspheres.
UPLC;HPLC;Gatifloxacin microspheres;Related substances;Light stability
R927.2
A
1001-0408(2012)33-3127-03
DOI10.6039/j.issn.1001-0408.2012.33.21
Δ廣東省醫學科研基金項目(A2010296)
*高級工程師。研究方向:藥物新劑型和質量標準。電話:020-39352507。E-mail:dengh361@sohu.com
#通訊作者:教授,碩士。研究方向:藥物新劑型。電話:020-39352507。E-mail:linzhangshu@tom.com
2012-03-20
2012-05-09)
