凝膠注模的凝膠反應動力學和均勻性
2012-11-29 10:33:48王小鋒王日初彭超群羅玉林王志勇
中南大學學報(自然科學版) 2012年4期
王小鋒,王日初,彭超群,羅玉林,王志勇
(1.中南大學 材料科學與工程學院,湖南 長沙,410083;2.中南大學 冶金科學與工程學院,湖南 長沙,410083)
在過去的幾十年中,由于能夠制備形狀復雜且微觀組織結構均勻的坯體,陶瓷膠態成型技術一直是研究的熱點[1?4]。凝膠注模是一種重要的原位凝固膠態成型技術。除具有可成型復雜形狀部件和坯體結構均勻等普通膠態成型的優點之外,它還具有坯體有機物含量低且強度高、工藝過程易控制和成本低廉等一系列優點[5]。該技術已用于 Al2O3[5,6],ZrO2[7],SiC[8]和 PZT[9]等各種氧化物或非氧化物陶瓷材料體系。目前,采用丙烯酰胺凝膠體系的水基凝膠注模因過程易控和適用性廣等特點受到廣泛關注[5?9],其原理為利用丙烯酰胺單體在懸浮液中進行凝膠反應形成具有三維網絡結構的高分子物質,將陶瓷粉體顆粒原位固定,從而獲得具有所需形狀要求的制品。在凝膠注模的工藝過程中,懸浮液的原位固化即丙烯酰胺單體通過凝膠反應形成網絡結構高分子的過程是十分重要的一道工序。Omatete等[6]研究懸浮液溫度隨凝膠反應時間的變化,并且指出凝膠反應的誘導期和凝膠點即凝膠反應開始發生的時間。馬利國等[10]研究凝膠注模的固化過程,并分析陶瓷漿料凝膠點的影響因素。仝建峰等[11]研究表明可通過建立懸浮液溫度與時間的曲線關系來研究懸浮液的凝固動力學。戴春雷等[12]甚至還研究凝膠注模的延遲固化。但上述研究均是以懸浮液中的凝膠反應為研究對象進行的。實際上,因為懸浮液的變化如陶瓷粉體材料體系的改變、粘度的變化和起皮抑制劑如聚乙二醇[13]的加入等對凝膠反應、坯體強度和均勻性等都會產生影響,所以,水溶液中的凝膠反應和凝膠均勻性才是凝膠注模的基礎,很有必要對其進行研究。本文作者研究各工藝條件如引發劑濃度、催化劑濃度、單體濃度、單體與交聯劑的質量比和起始反應溫度等對凝膠反應和凝膠均勻性的影響。此外,還研究懸浮液固相體積分數對凝膠反應的影響。
1 實驗材料與方法
1.1 原料
選用湖南水口山有色金屬集團有限公司生產的氧化鈹粉體為原料,平均粒徑為0.5 μm。有機單體和交聯劑分別為丙烯酰胺(AM)和 N,N’-亞甲基雙丙烯酰胺(MBAM)(上海國藥集團有限公司);引發劑為過硫酸銨(APS,長沙分路口化工廠);催化劑為N,N,N',N'-四甲基乙二銨(TEMED,北京化學試劑公司);分散劑為聚丙烯酸銨(日本東亞合成有限公司);去離子水為實驗室自制水。
1.2 實驗過程
精確稱取APS配制質量分數為10%的溶液待用。稱取一定量的單體AM和交聯劑MBAM加入去離子水中,經攪拌均勻直至溶液透明,得到單體水溶液。在單體水溶液中加入分散劑NH4PAA和BeO粉體,球磨24 h制得固相體積分數為45%的BeO粉體懸浮液。
在單體水溶液中加入一定量的催化劑TEMED和引發劑APS溶液,靜置;該溶液經一定時間反應后轉變為凝膠。為了排除因氧氣存在而產生“氧阻聚”的影響,將溶液在反應前抽真空處理,除去其中的氧氣。
將BeO粉體懸浮液真空除氣后,在設定溫度下加入引發劑和催化劑并注入模具中,使懸浮液凝膠固化。
1.3 測試與表征
丙烯酰胺單體的自由基聚合過程是一個放熱反應,其聚合熱為82.8 kJ/mol[14],放熱引起的溫度變化比較明顯。因此,采用聚合反應的熱效應,即溫度變化來表征單體聚合的轉化率和聚合速度[12]。將一定量的單體水溶液或懸浮液加入燒杯內,然后將之置于可控溫的保溫容器內,待溫度達到設定的溫度后,加入引發劑APS或催化劑TEMED引發反應。同時,將溫度探頭插入反應溶液中,監測并記錄反應體系溫度隨時間的變化。
將合成后的凝膠浸泡至溶脹平衡,切成長×寬×高為20 mm×10 mm×5 mm的小塊,置于40 mm×10 mm×5 mm的比色皿中,再注入蒸餾水。用721B型可見分光光度計,在波長為580 nm時測定凝膠的透射比[15]。
2 實驗結果
2.1 引發劑體積分數對凝膠反應和凝膠透射比的影響

圖1 引發劑體積分數對凝膠反應的影響Fig.1 Effect of initiator concentration on gelation
圖1所示為不同引發劑體積分數時凝膠反應的溫度變化和對應的誘導期與反應期。由圖1可知:隨著引發劑體積分數的增加,凝膠反應誘導期持續縮短。此外,凝膠反應“S”曲線的斜率也逐漸增加,表明聚合速率也升高,因此,反應期也縮短。
圖2所示為引發劑體積分數對合成凝膠透射比的影響。由圖2可知:隨引發劑體積分數的增加,凝膠透射比先增加而后減小,但總的來說引發劑體積分數對合成凝膠透射比的影響不大。
2.2 催化劑體積分數對凝膠反應和透射比的影響
圖3所示為不同催化劑體積分數時凝膠反應的溫度變化和相應的誘導期與反應期。由圖3可知:隨著催化劑體積分數的增加,誘導期和反應期均減小。
圖4所示為催化劑體積分數對凝膠透射比的影響。由圖4可知:隨著催化劑體積分數的增加,凝膠透射比整體上逐漸減小,但總的來說對合成凝膠透射比的影響也不大。
2.3 單體質量分數對凝膠反應和透射比的影響
圖5所示為單體質量分數對凝膠反應的影響和對應的誘導期與反應期。由圖5可知:隨著單體質量分數的增加,曲線的斜率也逐漸增加,表明增加單體質量分數能夠促進聚合反應,使聚合反應速率增加。此外,各反應的最高溫度也基本隨單體質量分數的增加而增加。單體質量分數的增加使得參與反應的乙烯基濃度增加,而該反應為一個放熱反應,因此,反應的最高溫度也必然增加。反應期和誘導期隨單體質量分數的增加而縮短(如圖5(b)所示),即單體質量分數的增加提高了引發速率。
圖6所示為單體質量分數對凝膠透射比的影響。由圖6可見:單體質量分數過高或過低,凝膠的透明性均會降低,表明凝膠的結構均勻性變差。

圖2 引發劑體積分數對合成凝膠透射比的影響Fig.2 Effect of initiator concentration on transmittance of gel

圖3 催化劑體積分數對凝膠反應的影響Fig.3 Effect of catalyst concentration on gelation

圖4 催化劑體積分數對凝膠透射比的影響Fig.4 Effect of catalyst concentration on transmittance of gel
2.4 單體與交聯劑質量比對凝膠反應和透射比的影響

圖5 單體質量分數對凝膠反應的影響(引發劑和催化劑體積分數分別為6 μL/100 mL和 3 μL/100 mL)Fig.5 Effect of monomer concentration on gelation

圖6 單體質量分數對合成凝膠的透射比的影響Fig.6 Effect of monomer concentration on transmittance of gel
圖7所示為單體比例對凝膠反應的影響和對應的誘導期與反應期。由圖7可知:隨著單體與交聯劑質量比(m(AM)/m(MBAM))比例增加,凝膠反應的誘導期和反應期均縮短。圖7還表明:凝膠聚合反應速率也體現在反應熱效應(最高溫度)上。當m(AM)/m(MBAM)為10∶1時,聚合反應放熱產生的熱積累使溶液的溫度升高23.6 ℃;而當m(AM)/m(MBAM)為35∶1時,溶液的溫度升高20.2 ℃,兩者溫度相差3.4 ℃。此外,圖7中各“S”曲線中間段的斜率均十分相近,也表明在不同m(AM)/m(MBAM)的條件下,凝膠聚合反應速率相差不大。另外,與單體質量分的影響(圖5)比較,m(AM)/m(MBAM)對凝膠反應的影響比單體濃度的影響小許多。顯然,m(AM)/m(MBAM)對反應誘導期和反應期的影響也不大(如圖7(b)所示)。

圖7 m(AM)/m(MBAM)對凝膠反應的影響Fig.7 Effect of m(AM)/m(MBAM)ratio on gelation
圖8所示為不同m(AM)/m(MBAM)下合成凝膠的透射比。由圖8可以看出:隨著m(AM)/m(MBAM)的降低即交聯劑濃度的增加,凝膠透明性有所降低。由圖8還可知:交聯劑濃度對凝膠透射比的影響遠大于單體AM濃度的影響因素,而且在單體比例為10∶1與15∶1之間(交聯劑濃度分別為0.097和0.065 mol/L)存在突變。

圖8 m(AM)/m(MBAM)對合成凝膠的透射比的影響Fig.8 Effect of m(AM)/m(MBAM)ratio on transmittance of gel
2.5 起始反應溫度對凝膠反應和透射比的影響
圖9所示為起始反應溫度對凝膠反應的影響和對應的誘導期與反應期。從圖9可見:隨起始反應溫度的增加,凝膠反應的誘導期和反應期均縮短;隨起始反應溫度的增加,反應從開始到結束的時間即持續的時間逐漸減少。在起始反應溫度為40 ℃的條件下,反應持續的時間僅為30 s左右。
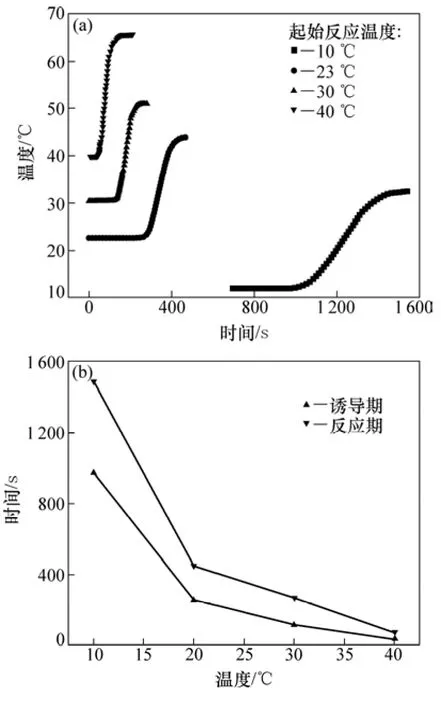
圖9 起始反應溫度對凝膠反應的影響(引發劑濃度和催化劑體積分數分別為6 μL/100 mL和2 μL/100 mL)Fig.9 Effect of initial reaction temperature on gelation
圖10 所示為在不同起始反應溫度條件下制備凝膠的透射比。由圖10可知:隨起始反應溫度的增加,透射比先增加而后又小幅度下降。
2.6 固相體積分數對凝膠反應的影響

圖10 起始反應溫度對凝膠透射比的影響Fig.10 Effect of initial reaction temperature on transmittance of gel

圖11 固相體積分數對凝膠反應的影響Fig.11 Effect of solids loading on gelation
圖11所示為不同固相體積分數的懸浮液中凝膠反應體系溫度隨時間的變化曲線。從圖11可見:懸浮液固相體積分數對凝膠反應的影響很大;隨固相體積分數的增加,凝膠反應誘導期縮短,而反應期卻小幅度延長(如圖11(b)所示),表明凝膠反應的引發速率升高。
3 討論
3.1 凝膠反應的基本原理
凝膠注模的工藝過程為:首先通過球磨和超聲振蕩等方式配成含有機單體AM和交聯劑MBAM的高固相體積分數的粉體懸浮漿料,然后加入引發劑與催化劑等可使有機單體發生凝膠反應的物質,充分攪拌均勻后,將懸浮液注入模具中,最后在一定的溫度條件下經過一段時間后凝膠反應被引發,AM與MBAM相互反應形成具有一定強度和柔韌性的三維網狀結構,從而獲得具有一定形狀的坯體。凝膠注模的核心原理是采用形成的具有三維網絡結構的高分子物質將分散均勻的粉體懸浮液中的顆粒包裹使之原位固定。顯然,在整個工藝過程中,三維網絡結構高分子的形成,即懸浮液中AM與MBAM的凝膠反應,具有非常關鍵的作用。根據高分子原理,AM與MBAM之間的反應為典型的自由基聚合反應。一般地,自由基聚合反應分為鏈引發、鏈增長和鏈終止3步基元反應[14,16]。鏈引發:鏈引發是形成單體自由基的反應。本研究采用引發劑過硫酸銨(APS)和催化劑(TEMED)引發反應。鏈引發分為2步反應:首先水溶液中的APS在熱作用下均裂分解形成具有很高活性的初級自由基;然后它分別將AM和MBAM分子中的烯鍵打開并與之加成,形成單體自由基。鏈增長:所得單體自由基打開烯類分子的π鍵,加成,形成新自由基。新自由基的活性并不衰減,繼續與烯類單體連鎖加成,形成結構單元更多的鏈自由基。鏈終止:隨著反應的進行,水溶液中的自由基濃度迅速增加。高活性的自由基彼此間易相互作用而終止,形成穩定的聚合物分子。
3.2 凝膠反應動力學
凝膠反應過程中,因為單體聚合時會放出能量,從而引起反應體系溫度的升高,所以整個過程中溫度隨時間的增加而升高。忽略環境與反應體系間的傳熱關系,可采用反應體系溫度隨時間的變化關系來表征凝膠反應速率的變化[12,16]。在自由基聚合的反應中,鏈引發是最慢的一步,控制著總的聚合速率。引發劑濃度是影響速率和分子量的關鍵因素,對凝膠反應有很大影響。引發劑分解后,一部分用來引發單體聚合,另一部分則因誘導分解或籠蔽效應而消耗。誘導分解是一種自由基向引發劑的轉移反應。籠蔽效應則是處在單體或溶劑“籠子”中的引發劑分子無法及時擴散出來,在籠內發生副反應形成穩定分子而消耗的現象。當引發劑濃度較低時,由于受到誘導分解和籠蔽效應的影響,引發劑分解產生的自由基很容易被損耗,沒有足夠多的引發劑自由基和單體作用形成單體自由基,進而進行鏈增長反應。隨引發劑濃度的增加,引發劑自由基增多,單體自由基也隨之增加,凝膠反應能夠被完全引發,因此誘導期也隨著引發劑濃度的增加而縮短,如圖5(b)所示。并且增加引發劑濃度的效果十分明顯,因此圖5(b)中曲線較陡峭。當引發劑達到一定濃度后,產生的自由基足夠且遠遠大于鏈增長反應的所需。因此,多余的自由基并沒有參加鏈增長反應,相應地就不能更有效地縮短誘導期和反應期,表現在圖5(b)中為曲線趨于平緩。
催化劑在反應過程中起到加速自由基產生和降低反應活化能的作用,因此,催化劑的加入必然使誘導期和反應期縮短(圖3)。另外,催化劑濃度較小時,其作用十分明顯;而催化劑的加入量已經較多時,其起到的加速作用則不再明顯。凝膠反應中對誘導期和反應期起主要作用是溶液內自由基的量。少量的催化劑與引發劑反應能降低其生產自由基的激活能,因此,作用明顯;當催化劑過量時,由于引發劑已幾乎被全部消耗,自由基產生的量不再增加,過量的催化劑所起的作用沒那么明顯。
在某些情況下,除引發劑濃度外,單體濃度對鏈引發速率也有影響。單體濃度越高,初級自由基與單體結合進行鏈引發的概率也就越高,凝膠反應發生得越快。單體濃度與引發速率存在如下關系[16]:

相應地,與聚合速率存在如下關系:

式中:Ri,Rp,I,M,kd,kt和f分別代表反應速率、增長速率、引發劑濃度、單體濃度、引發速率常數、終止速率常數和引發劑效率。
式(1)和式(2)表明:增加單體濃度,引發速率和聚合速率均相應地升高。因此,凝膠反應的誘導期和反應期均縮短(圖5)。
單體與交聯劑質量比對凝膠反應也有影響。與單體濃度的影響原因相同,m(AM)/m(MBAM)增加,即MBAM濃度下降,使得溶液中的乙烯基濃度降低,因此,聚合反應速率稍微降低,凝膠反應的誘導期和反應期均縮短。
起始反應溫度對凝膠反應起著十分重要的作用。Gelfi等[17]的研究表明:只有當起始反應溫度高于某一溫度之后,凝膠反應才能進行。起始反應溫度的增加使得引發劑分解速率增加,自由基濃度升高,引發速率增加,因此,凝膠反應的誘導期縮短(圖9)。
引發劑分解產生自由基是一個熱激活的過程,滿足阿倫尼烏斯方程:

其中:r為引發速率,R為氣體參數,Ea為引發劑分解產生自由基的激活能。因此,采用圖9中的數據,根據方程(3)可計算引發劑分解激活能,如圖12 所示。從圖12可見:反應激活能為79.45 kJ/mol,該值與其他文獻報道的結果基本一致[6]。

圖12 引發劑分解激活能的計算Fig.12 Calculation of active energy of decomposition of initiator
增加懸浮液的固相體積分數能促進凝膠反應的進行。凝膠反應是靠溶液中的自由基引發而進行,并且自由基濃度直接決定凝膠反應速率。引發劑形成的自由基,因懸浮液黏度較高而無法快速擴散,從而造成局部引發劑濃度過高,引發速率增加。而聚丙烯酰胺凝膠反應為鏈式反應,即一旦被引發后將持續進行直至反應完成。所以,局部過高的自由基濃度將造成整個反應引發速率升高并持續進行到底。顯然,懸浮液固相體積分數越高,黏度越大,反應引發效率越高,具體表現為反應誘導期越短(如圖11(b)所示)。對于固相體積分數為20%的懸浮液而言,其凝膠反應的誘導期為340 s,而當固相體積分數增至50%時,其凝膠反應的誘導期縮短到190 s。此外,懸浮液的高黏度還會使得聚合反應進行緩慢,聚合速率下降,因此,凝膠反應的反應期延長(如圖11(b)所示)。固相體積分數為50%的懸浮液,其反應期長達650 s,且在反應后期,反應體系的溫度持續緩慢上升。對比圖11(a)中各曲線還可知:隨固相體積分數的增加,各“S”曲線中間部分的斜率逐漸降低,也表明凝膠反應的聚合速率逐漸下降;另外,反應體系的最高溫度也隨固相體積分數的增加而下降。當固相體積分數為20%時,反應體系的最高溫度為36.6 ℃,而當固相體積分數為50%時,最高溫度降低到30 ℃。固相體積分數的增加使凝膠反應體系的比熱容也相應地增加,而聚合反應放出的能量一定,因此,反應體系的最高溫度必然下降。
3.3 凝膠的均勻性
坯體結構均勻是凝膠注模的眾多優點之一[5]。但如果凝膠注模過程控制不當,其優點就不能很好地體現出來,甚至有可能會成為缺點。顯然,凝膠注模坯體為凝膠和粉體的復合體,因此,坯體結構的均勻性很大程度上受凝膠均勻性的影響,很有必要對凝膠的均勻性進行研究和討論。
凝膠化學[18]認為凝膠形成過程是支化結構在整個體系內擴張并占據體系分攤整個空間的過程。但支化結構的發展并不是從體系的一點開始的,而是多個出發點同時開始生長并相互穿過邊界而結合,最終鏈終止生長,獲得凝膠物質。顯然,凝膠的非均質性與支化結構的數量和分布有很大關系,而支化結構受凝膠反應條件如引發劑、催化劑、單體和交聯劑等的濃度影響,特別與單體和交聯劑的反應活性即競聚率有關。凝膠反應條件影響凝膠反應速率和分子量即支化結構空間占有量。競聚率不同使交聯反應可能優先發生,或者最后發生,使交聯點在聚合物中集中于某些特定部分,這些交聯點密集的部位稱為凝膠中的微凝膠。若這些微凝膠在宏觀凝膠中分布不均勻,則成為非均質凝膠。在可見光波長范圍內,由于光通過凝膠時產生散射,因而非均質凝膠是不透明的,而交聯點均勻分布的凝膠,由于沒有折射率的差別,因此,通常光照時是透明的。因此,可采用可見光分光光度計測試凝膠在某一波長的透射比來評價凝膠的均勻性。
凝膠的均勻性取決于其網絡均勻性和結構均勻性,因此,從凝膠微觀組織結構的角度來看,凝膠分為如下3種:均質網絡均質結構凝膠、非均質網絡非均質結構凝膠和均質網絡非均質結構凝膠。圖13所示為各種凝膠的網絡結構示意圖[18]。圖13(a)所示的網絡是交聯點間分子量全部相等和交聯點在空間分布均勻的均質網絡鏈,稱為“理想網絡”,常作為理論模型使用。圖13(b)所示的網絡是由于交聯點之間相對分子質量不同,交聯空間分布不均勻的凝膠網絡,實際上大多凝膠是這種結構。圖13(c)的情況與圖13(a)的一樣,無分布、交聯點間分子量相等,但是,交聯點的空間位置不均勻,是非均質狀態凝膠。

圖13 3種不同凝膠的網絡結構示意圖[18]Fig.13 Schematic of three different gels network structure[18]
顯然,通過控制凝膠反應條件如引發劑濃度、催化劑濃度、單體濃度、交聯劑濃度和起始反應溫度等,可獲得具有網絡較均勻且結構較均勻的凝膠,從而提高凝膠均勻性。網絡均勻性主要是通過控制引發劑濃度和催化劑濃度等條件來實現。當引發劑和催化劑濃度較低時,凝膠反應引發速率較低,聚合速率也較小,單體的轉化率較低,因此,聚合物的分子鏈長短不一,網絡均勻性較差,凝膠均勻性不好。隨引發劑和催化劑濃度的增加,反應引發速率和聚合速率逐漸增加,轉化率升高,網絡均勻性增加,凝膠的均勻性也增加。而當引發劑濃度較高時,反應引發速率和聚合速率急劇增加,此時,雖然單體轉化率較高,但聚合反應所得產物的相對分子質量急劇下降,同時可能存在局部自由基濃度較高而出現“暴聚”的現象。所以,在引發劑和催化劑濃度較高條件下所得凝膠的網絡結構均勻性下降,凝膠均勻性反而降低。但總體來說,引發劑和催化劑濃度對凝膠均勻性的影響有限。與單體濃度和m(AM)/m(MBAM)等反應條件相比較,引發劑和催化劑的加入只改變了凝膠反應速度,對最終凝膠微觀組織的影響不大,相應地,對其均勻性的影響也不是很大(圖2和圖4)。
凝膠反應條件如單體濃度和交聯劑濃度等不僅對凝膠網絡均勻性,而且對結構均勻性有較大的影響。如前所述,單體濃度對聚合速率影響很大(圖5)。當單體濃度較低時,引發速率過低,不利于單體聚合,所得凝膠的分子鏈不均勻,因此,其網絡均勻性較差(圖6);而當單體濃度較高時,其與交聯劑進行交聯形成支化結構的概率增加,因此結構均勻性變差,凝膠均勻性下降(圖6)。此外,單體濃度對凝膠透明性的影響還可以歸因于聚合熱效應累積導致相分離,從而得到不均勻的混濁凝膠。因此,在凝膠注模的工藝過程中,須選擇恰當的單體濃度,其較合適的范圍(質量分數)為 10%~20%(圖6)。交聯劑濃度(單體交/聯劑比例)對凝膠均勻性存在很大的影響(圖8)。隨交聯劑濃度的增加即單體交/聯劑比例的降低,其與單體進行交聯形成的支化結構增加,所以凝膠的結構均勻性變差,凝膠均勻性下降。當交聯劑濃度大于某一值時,所形成的水凝膠不透明。這可能是AM與MBAM在水中溶解度差別較大,隨著m(AM)/m(MBAM)的降低,MBAM會聚集形成許多束狀區域,這種聚集束的 MBAM 經聚合可形成高密度的聚合物,并達到可見光波長大小的鏈束,因而水凝膠透明性降低。這與 Gelfi等的研究[19]結果一致。此外,MBAM在水溶液中的溶解度恰好落在突變范圍內。這進一步證實了凝膠透明性的變化是由 MBAM 溶解度的變化引起的。綜上所述,為了獲得具有均勻網絡結構的透明凝膠,m(AM)/m(MBAM)應高于 20∶1(圖8)。
起始反應溫度對溶解度存在影響,所以,當起始反應溫度較低時(如10 ℃),因交聯劑MBAM在溶液中的溶解度較低而部分未溶,所以,合成的凝膠結構不均勻,呈乳白色。當起始反應溫度較高時(如40 ℃),凝膠反應十分迅速,容易造成局部反應不均,凝膠網絡均勻性下降,因此,凝膠透射比稍微降低。Gelfi等[17]研究溫度對凝膠反應動力學的影響時發現:溫度對凝膠均勻性存在很大影響,過低的溫度甚至可使凝膠反應無法進行。因此,凝膠注模的起始反應溫度也應控制在20~40℃(圖10)。
4 結論
(1)引發劑濃度和催化劑濃度對凝膠反應速率有很大影響,但對凝膠均勻性的影響不大。隨引發劑和催化劑濃度的增加,凝膠反應誘導期和反應期均迅速縮短。引發劑和催化劑濃度過高或過低均不利于凝膠反應的進行,影響凝膠微觀組織結構的網絡均勻性,因此,凝膠均勻性下降。可見,引發劑和催化劑濃度應控制適中。
(2)增加單體濃度,凝膠反應引發速率和聚合速率均相應地升高,因此,誘導期和反應期均縮短。單體濃度對凝膠均勻性有影響。當單體濃度較低時,所得凝膠的分子鏈不均勻,因此,其網絡均勻性較差;當單體濃度較高時,其與交聯劑形成的支化結構增加導致凝膠的結構均勻性變差,同時,聚合熱效應累積增加導致相分離,所以,凝膠均勻性下降。故在凝膠注模的工藝過程中,單體質量分數較合適的范圍為10%~20%。
(3)單體與交聯劑質量比對凝膠反應以及誘導期和反應期影響不大,但對凝膠均勻性的影響很大。隨著單體交與聯劑質量比的降低,形成的支化結構增加,所以凝膠的結構均勻性變差,凝膠均勻性下降。當單體與交聯劑質量比低于某一值時,由于交聯劑溶解度較低而會聚集形成的束狀區域在聚合過程中形成高密度的聚合物,因此凝膠均勻性降低。所以,對于凝膠注模的均勻性而言,單體與交聯劑質量比應高于20∶1。
(4)起始反應溫度對凝膠反應和均勻性都有很大影響。當起始反應溫度較低時,因交聯劑 MBAM 在溶液中的溶解度較低,凝膠結構不均勻;當起始反應溫度較高時,過高的凝膠反應速度造成局部反應不均,形成的網絡均勻性下降,凝膠均勻性降低。因此,凝膠注模的起始反應溫度應控制在20~40 ℃。
(5)懸浮液固相體積分數的增加使得黏度升高,從而造成局部自由濃度過高,因此,能促進凝膠反應的進行,誘導期縮短。但懸浮液的高黏度還會使得聚合速率下降,因此,凝膠反應的反應期延長。
致謝:感謝天津大學的方道斌教授在實驗過程中給予的幫助。
[1]Lange F F.Powder processing science and technology for increased reliability[J].Journal of the American Ceramic Society,1989,72(1): 3?15.
[2]Baader F H,Graule T J,Gauckler L J.Direct coagulation casting:A new green shaping technique.Part I: Processing principles[J].Industrial Ceramics,1996,16(1): 31?36.
[3]Lewis J A.Colloidal processing of ceramics [J].Journal of the American Ceramic Society,2000,83(10): 2341?2359.
[4]Olhero S M,Ganesh I,Torres P M C,et al.Aqueous colloidal processing of ZTA composites[J].Journal of the American Ceramic Society,2009,92(1): 9?16.
[5]王小鋒,王日初,彭超群,等.凝膠注模成型技術的研究與進展[J].中國有色金屬學報,2010,20(3): 496?509.WANG Xiao-feng,WANG Ri-chu,PENG Chao-qun,et al.Research and development of gelcasting[J].The Chinese Journal of Nonferrous Metals,2010,20(3): 496?509.
[6]Omatete O O,Janney M A,Strehlow R A.Gelcasting: A new ceramic forming process[J].American Ceramic Society Bulletin,1991,70(10): 1641?1649.
[7]Adolfsson E.Gelcasting of zirconia using agarose [J].Journal of the American Ceramic Society,2006,89(6): 1897?1902.
[8]DONG Man-jiang,MAO Xiao-jian,ZHANG Zhao-quan,et al.Gelcasting of SiC using epoxy resin as gel former[J].Ceramic International,2009,35(4): 1363?1366.
[9]GUO Dong,CAI Kai,LI Long-tu,et al.Gelcasting of PZT[J].Ceramics International,2003,29(4): 403?406.
[10]馬利國,黃勇,楊金龍,等.凝膠注模成型固化過程及其影響因素: 陶瓷漿料凝膠點測定及其影響因素的研究[J].成都大學學報: 自然科學版,2002,21(2): 5?10.MA Li-guo,HUANG Yong,YANG Jin-long,et al.Solidification course and its influence factors for gelcasting: Gel point measurement of ceramics slurry and its influence factor[J].Journal of Chengdu University: Natural Science,2002,21(2):5?10.
[11]仝建峰,陳大明,李寶偉,等.氧化鋁陶瓷凝膠注模成型凝固動力學研究[J].航空材料學報,2008,28(3): 49?52.TONG Jian-feng,CHEN Da-ming,LI Bao-wei,et al.Solidification kinetics of alumina suspension by gelcasting [J].Journal of Aeronautical Materials,2008,28(3): 49?52.
[12]戴春雷,楊金龍,黃勇.凝膠注模成型延遲固化研究[J].無機材料學報,2005,20(1): 83?89.DAI Chun-lei,YANG Jin-long,HUANG Yong.Investigation on delay solidification for gelcasting[J].Journal of Inorganic Materials,2005,20(1): 83?89.
[13]LI Fei,CHEN Hai-yan,WU Rui-zhi,et al.Effect of polyethylene glycol on the surface exfoliation of SiC green bodies prepared by gelcasting[J].Materials Science and Engineering A,2004,368: 255?259.
[14]方道斌,郭睿威,哈潤華.丙烯酰胺聚合物[M].北京: 化學工業出版社,2006: 143?181.FANG Dao-bin,GUO Rui-wei,HA Run-hua.Polymers of acrylamide[M].Beijing: Chemical Industry Press,2006:143?181.
[15]張艷群,哈鴻飛.氯化鈉相轉變K-型卡拉膠/聚N-異丙基丙烯酰胺共混凝膠的輻射合成及性質研究[J].高分子學報,2001,33(4): 485?488.ZHANG Yan-qun,HA Hong-fei.Radiation synthesis and properties of Kappa-carrageenan-poly(N-isopropylacrylamide)hydrogel blends[J].Acta Polymerica Sinica,2001,33(4):485?488.
[16]潘祖仁.高分子化學[M].北京: 化學工業出版社,2006:78?80.PAN Zu-ren.Polymer Chemistry[M].Beijing: Chemical Industry Press,2006: 78?80.
[17]Gelfi C,Righetti P G.Polymerization kinetics of polyacrylamide gels II.Eeffect of temperature[J].Electrophoresis,1981,2(4):220?228.
[18]顧雪蓉,朱育平.凝膠化學[M].北京: 化學工業出版社,2004:57?117.GU Xue-rong,ZHU Yu-ping.Gel chemistry[M].Beijing:Chemical Industry Press,2004: 57?117.
[19]Gelfi C,Righetti P G.Polymerization kinetics of polyacrylamide gels I.Eeffect of different cross-linkers[J].Electrophoresis,1981,2(4): 213?219.
