飛秒時間分辨的光電子影像研究氯化芐分子內(nèi)轉(zhuǎn)換動力學(xué)
2012-12-11 09:31:00丁中華邱學(xué)軍徐晏琪王艷梅
物理化學(xué)學(xué)報 2012年12期
關(guān)鍵詞:物理
丁中華 邱學(xué)軍 徐晏琪 王艷梅 張 冰
(中國科學(xué)院武漢物理與數(shù)學(xué)研究所,波譜與原子分子物理國家重點實驗室,武漢430071;中國科學(xué)院大學(xué),北京100049)
飛秒時間分辨的光電子影像研究氯化芐分子內(nèi)轉(zhuǎn)換動力學(xué)
丁中華 邱學(xué)軍 徐晏琪 王艷梅*張 冰
(中國科學(xué)院武漢物理與數(shù)學(xué)研究所,波譜與原子分子物理國家重點實驗室,武漢430071;中國科學(xué)院大學(xué),北京100049)
結(jié)合時間分辨的飛秒光電子影像(TRPEI)技術(shù)和時間分辨的質(zhì)譜技術(shù),研究了氯化芐(BzCl)分子內(nèi)轉(zhuǎn)換動力學(xué)過程.從光電子影像中獲得了光電子動能分布和角度分布.氯化芐分子吸收兩個400 nm的光子后從基態(tài)躍遷到S4態(tài)和S2態(tài).獲得的母體離子隨泵浦-探測時間延遲變化的曲線可以用兩個指數(shù)函數(shù)進(jìn)行擬合,包括一個時間常數(shù)為50 fs的快速組分和一個時間常數(shù)為910 fs的慢速組分.通過分析光電子動能分布隨延遲時間的變化,我們認(rèn)為分子被激發(fā)到S4態(tài)后在很短的時間內(nèi)與S2態(tài)發(fā)生耦合迅速弛豫到S2態(tài),然后再經(jīng)內(nèi)轉(zhuǎn)換(IC)弛豫到S1態(tài).最初布居的激發(fā)態(tài)分子經(jīng)過內(nèi)轉(zhuǎn)換弛豫到S1態(tài)的時間尺度為50 fs.910 fs的慢速時間組分反映了分子弛豫到S1態(tài)后,經(jīng)內(nèi)轉(zhuǎn)換向基態(tài)S0的弛豫.光電子角度分布的各向異性參數(shù)從零時刻的0.87增加到25 fs時的0.94,然后逐漸減小到190 fs時刻的0.59的現(xiàn)象,也反映了氯化芐分子從S4態(tài)耦合到S2態(tài),然后內(nèi)轉(zhuǎn)換到S1態(tài)的動力學(xué)過程.
氯化芐;光電子影像;內(nèi)轉(zhuǎn)換;泵浦-探測;時間分辨光譜
1 Introduction
Ultrafast dynamics of electronically excited molecules plays an important role in photo-reaction,life science,and environmental science.The deactivation processes of electronically excited molecules in gas phase include internal conversion(IC), intersystem crossing(ISC),intramolecular vibrational energy redistribution(IVR),and so on.Of special interest are the mechanisms for IC1-3of electronically excited polyatomic molecules into vibrational modes of lower electronic levels and the ground state.IC,which has been widely studied,can be induced by conical intersection of potential energy surfaces in polyatomic molecules.4This ultrafast IC decay process plays an essential role in the photostability of polyatomic molecules, for example DNA.5Recently-developed femtosecond timeresolved photoelectron imaging(TRPEI)technique6-9is extremely useful to investigate excited state dynamics in isolated polyatomic molecules.Because it is sensitive to both electronic configurations and vibrational dynamics,TRPEI is well suited to the study of ultrafast non-adiabatic processes.10Compared to femtosecond time-resolved photoelectron spectroscopy,TRPEI has the advantage of measuring the time evolution of both energy and angular distributions simultaneously.11Associated with resonance-enhanced multiphoton ionization(REMPI),TRPEI allows for the study of electronic relaxation dynamics in isolated molecules,as well as the study of sequential ultrafast electronic processes in both optically bright and dark states.
Benzyl chloride(BzCl)has been frequently used as a radical source because of its high quantum yield to generate benzyl radicals and chlorine atoms when photoexcited by light irradiation.12-16A large number of investigations for photodissociation of BzCl have been reported both in gas phase14,15,17,18and in condensed phase.19-21The experimental results of Ichimura and Mori14showed that the dissociation of BzCl vapor photoexcited by 184.9,206.0,and 253.7 nm undergoes C―Cl bond dissociation to give benzyl(Bz)radicals and chlorine atoms(Cl).Recently,Nagano and coworkers17studied the reaction intermediates with photolysis of BzCl by using two-color laser excitation transient absorption and two-color laser excitation time-resolved thermal lensing measurement.They proposed two dissociation pathways for BzCl and one of which is direct dissociation from the excited states.In 2005,Ichimura and coworkers18reported another experiment to measure the mass-resolved resonance enhanced two-photon ionization spectra of jet-cooled BzCl.Some low-frequency vibronic bands were assigned around the S1-S0origin band in their work.
In the present paper,we shed a light on the excited state dynamics of BzCl using TRPEI combined with time-resolved mass spectroscopy.We calculated the transition energies and Franck-Condon factors of the different excited states of BzCl molecule.Then we move our eyes to the photoelectron kinetic energy distributions.The energy flow between the S4,S2,and S1states was investigated.In the last,the photoelectron angular distributions were given to support our conclusion.
2 Experimental and computational methods
2.1 Experimental measurement
The time-resolved photoelectron imaging setup used in this study which has been described elsewhere,22,23consists of an amplified Ti:sapphire femtosecond laser system,a molecular beam apparatus,a time-of-flight mass-spectrometer(TOF-MS), and a two-dimensional(2D)position sensitive detector.
The details of our femtosecond laser system have been described elsewhere.22,23Briefly,the seed beam was generated by a commercial Ti:sapphire oscillator pumped by a CW second harmonic of an Nd:YVO4 laser,and then amplified by an Nd: YLF pumped regenerative amplifier(Coherent Int.Legend-USP-LE)to generate a 1 kHz pulse train centered at 800 nm of 45 fs pulse width with maximum energy of 1 mJ·pulse-1.The second harmonic pulse(400 nm)was generated in a 0.5 mm thick BBO crystal.The pump and probe beams with vertical polarization were merged by a dichroic mirror and directed into the molecular beam chamber.The time delay between the pump and probe pulses was accurately monitored by a computercontrolled linear translation stage(PI,M-126.CG1).
The liquid sample(benzyl chloride,99.9%purity),5%seeded in helium buffer gas at a background pressure of 200 kPa, was expanded through a pulsed valve to generate a pulsed molecular beam.The beam was skimmed and introduced into the ionization chamber where it was intersected perpendicularly with the laser beam.The electrons thus produced were accelerated parallel to the molecular beam and were then projected onto a position-sensitive imaging detector.The field-free region (360 mm)of the TOF spectrometer was shielded with aμ-metal tube to avoid external magnetic fields that might otherwise deflect the electron trajectories.The imaging detector consists of a micro channel plate(MCP)(Photek,VID-40),a phosphor screen,and an intensified charge-coupled device(CCD)camera(LAVISION,Imager Intense).In order to observe the total photoelectron and ion intensity,the emission from the phosphor screen was monitored with a photomultiplier tube(PMT).
2.2 Computational methods
Quantum chemical calculations on the molecules were performed in their ground states to dope out the optimized geometries of benzyl chloride at the Hartree-Fock(HF)/3-21g level of theory using Gaussian 09 suite of program.24Excitation energies to the excited electronic states were determined by time-dependent density functional theory(TD-DFT)with several different functionals and the 6-311++G basis set.
3 Results and discussion
Absorption spectra of benzyl chloride have been reported in the other paper.25Two bands corresponding to B2←A1and A1←A1electronic transitions have been observed.Here,ab initio computations were employed to get the transition energy of othersingletexcite states.The excitation energiesand Franck-Condon factors for transitions from electronic ground state are summarized in Table 1.Experimental data are also given for comparison.The theoretical values calculated by BPV86 and HCTH functionals are close to the experimental data,while the values calculated by B3LVP and B3PW91 have the larger error.It is visible that several excited singlet states lie below the energy of 6.2 eV.The S4and S2states which have the larger Franck-Condon factor are expected to have the largest probability to be populated.Thus benzyl chloride molecule may transit to the S4and S2states when excited by two photons of 400 nm.
In this experiment,the second harmonic pulse(400 nm)was used as the pump light and the fundamental pulse(800 nm)as the probe light.Their polarization regulated parallel to the detector.The mass spectra of benzyl chloride have three peaks corresponding to BzCl+,Bz+,and CH2Cl+,respectively.It is observed that the time profiles of photofragments are very similar to that of the parent ion.We attribute the photofragments to coming from the following dissociation of the parent ion. Therefore,they have no contributions to total photoelectron signal.The time-resolved total signal of the parent ion formed after two-photon excitation at 400 nm is shown in Fig.1 as a function of the pump-probe delay.The profile gives a biexponential decay with lifetimes of τ1=50 fs and τ2=910 fs convoluted with a Gaussian instrument response function.The time evolution will be discussed later by energy analysis of the corresponding photoelectron images.
The photoelectrons generated by pump-probe photoinization are projected onto a 2D position-sensitive detector.Photoelectron images are acquired as a function of the pump-probe time delay.Fig.2 shows reconstructed photoelectron images which represent the photoelectron kinetic energy distributions corresponding to such a sequence,inverted by the basis-set expansion method(BASEX).26Fig.3 shows the photoelectron spectrum extracted from the image acquired at the delay time of 50 fs.Three well-resolved bands which are assigned as the first, second,and third bands centered at about 0,0.61,and 0.98 eV, respectively,are observed.The maximum electron energy for different ionization scheme can be given by:
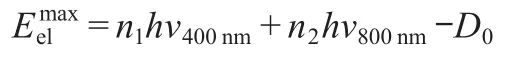
where the n1and n2represent photon numbers involved for 400 and 800 nm,respectively.D0is the adiabatic ionization potential of benzyl chloride.The total energy for(2+3?)ionization scheme,which means n1=2 and n2=3,would be 10.85 eV.Considering the ionization energy of benzyl chloride is 9.14 eV,27the maximum electron energy is 1.75 eV.Assuming that the energy of the excited S2state(5.18 eV)is conserved on ionization,one peak at about 0.69 eV would be expected.We ascribe the photoelectron kinetic energy(PKE)peak located at 0.61 eV to the D0←S2ionization process by three-photon at 800 nm with a small energy difference about 0.08 eV with the expected energy.Consider the bandwidth of the pump and probe laser(6 nm FWHM(full width at half maximum)for 400 nm and 30 nm FWHM for 800 nm),the error between the measured PKE and expected energy is considered acceptable under this condition.For the same reason the first band must arise from the S1state.The energy difference between the second and third bands is 0.37 eV which is in consistence of the calculated transition energy difference between the S2and S4states.For this reason we attribute the third band to arising from the ionization of the S4state by three photons of the probe pulse.
To gain more information about the time evolution of these four bands,Fig.4 shows the time-resolved PKE distributions extracted from a series of images.The time-resolved PKE distributions are characterized by a rapid decay of electron from the second and third bands and a growth of electron from the first band as the delay changes.The photoelectron spectrum shift from an energy band to another is a good signature of electronic configurations changing.According to the origin of these three bands,the energy shift from the second and third bands to the first is attributed to the internal conversion from the S4and S2states to S1state.
According to analysis above,the dynamics of benzyl chloride excited states have been studied,as shown in Fig.5.The excited S4state is initially populated following the absorption of two photons at 400 nm.Photoelectrons from the ionizationof S2state was also observed in the photoelectron spectra.The second and third bands which arise from S2and S4states respectively have same decay trend.This indicates that after initial promotion to the S4state,a very fast coupling between S4and S2states happened in a short time which cannot be resolved in this experiment.We cannot indentify whether the S3state involved in this process because the peak ionized from the S3state was not resolved in the PKE distributions.The probably reason for this is that the energy of S3state is very close to the S2state which causes that the electron coming from the S3state cannot be well separated from that coming from the S2state. The decay of S4and S2states may happen through an ultrafast internal conversion to S1at a time scale of 50 fs.Once decay to S1state,it is most likely that molecules will degenerate to S0state within 910 fs further.It can be seen from the Fig.1 that the signal from the S1state is very weak.It is maybe because the ionization probability from the S1state is so small that only a few molecules at S1state were ionized by the probe pulse.

Table 1 Calculated excitation energies(in eV)and Franck-Condon factors(f)of benzyl chloride
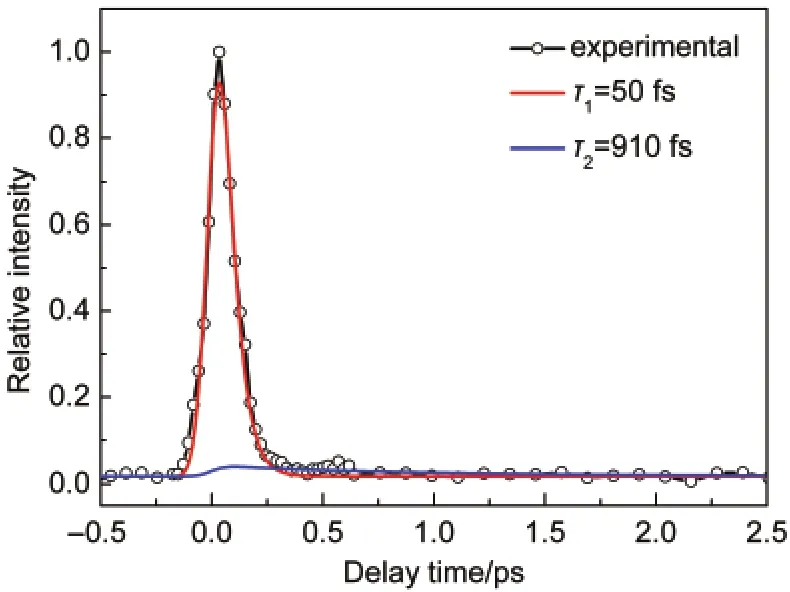
Fig.1 Time-resolved time of flight mass spectroscopy(TOF-MS) probing benzyl chloride as a function of delay timeThe circles represent the experimental results,the solid red and blue lines represent the fitted results.

Fig.2 Aseries of time-resolved BASEX-inverted photoelectron images result on benzyl chloride obtained by(2+3?)REMPI using λpump=400 nm and λprobe=800 nmThe linear polarizations of the pump and probe lasers are aligned vertical in the plane of the figure.
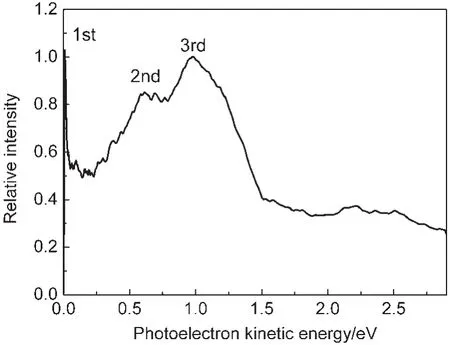
Fig.3 PKE distributions extracted from the image at a delay time of 50 fs
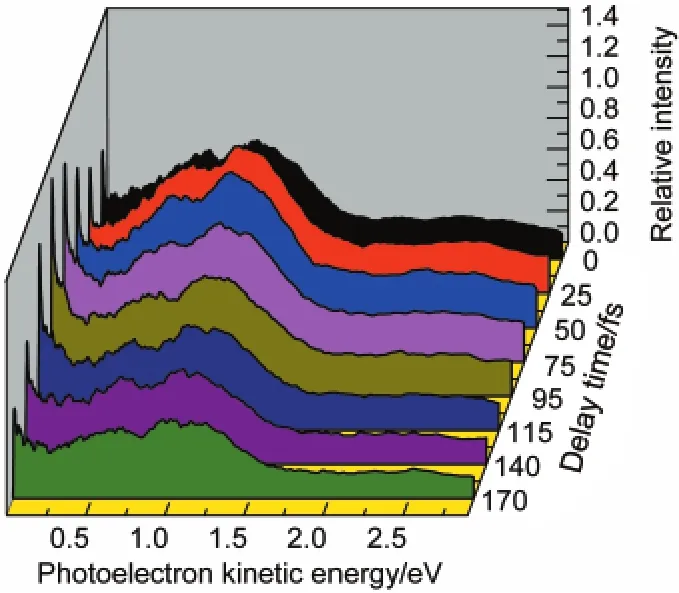
Fig.4 Time-energy distributions of the photoelectron intensity extracted from the images of Fig.2
In our experimental configuration,in which the pump and probe laser are linear and parallel to each other,the laboratory frame photoelectron angular distributions(PADs)resulting from ionization can be expanded as:28
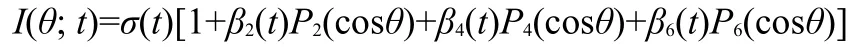
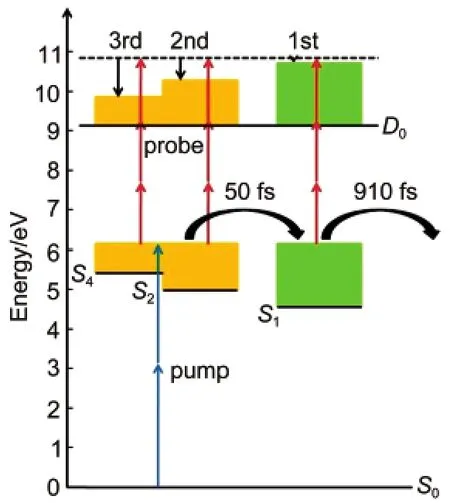
Fig.5 Schematic description of the photoionization of benzyl chloride
where σ(t)is the integral cross section,βn(t)are the nth order anisotropy parameters,Pn(cosθ)are the nth order Legendre polynomials and θ is defined as the angle between the laser polarization and velocity vector of the photoelectron.The anisotropy parameter β2is obtained for different photoelectron bands at different delay time. NosignificantchangesofPADsofthefirstband(withPKEdistributions near 0 eV)at different time delay are observed,which verifies no existence of rotational coherence in our experiment.PADs with distributions of PKE of 0.39-1.49 eV which contains the second and third bands are displayed in Fig.6.The value of β2changes from 0.87 to 0.94 at the first 25 fs.This may be caused by the rapid coupling effect from the S4to S2state at the first 25 fs.When excited by the pump pulse,the molecules were initially populated to S4and S2states.As the molecules decayed to S2state from the S4state,the ratio between the S4and S2states changed,which led to the variation of the value of β2.Then the IC from the S2state to the S1state changed the ratio between the S4and S2states further,which caused thatthevalueofβ2graduallychangedto0.59at190fs.
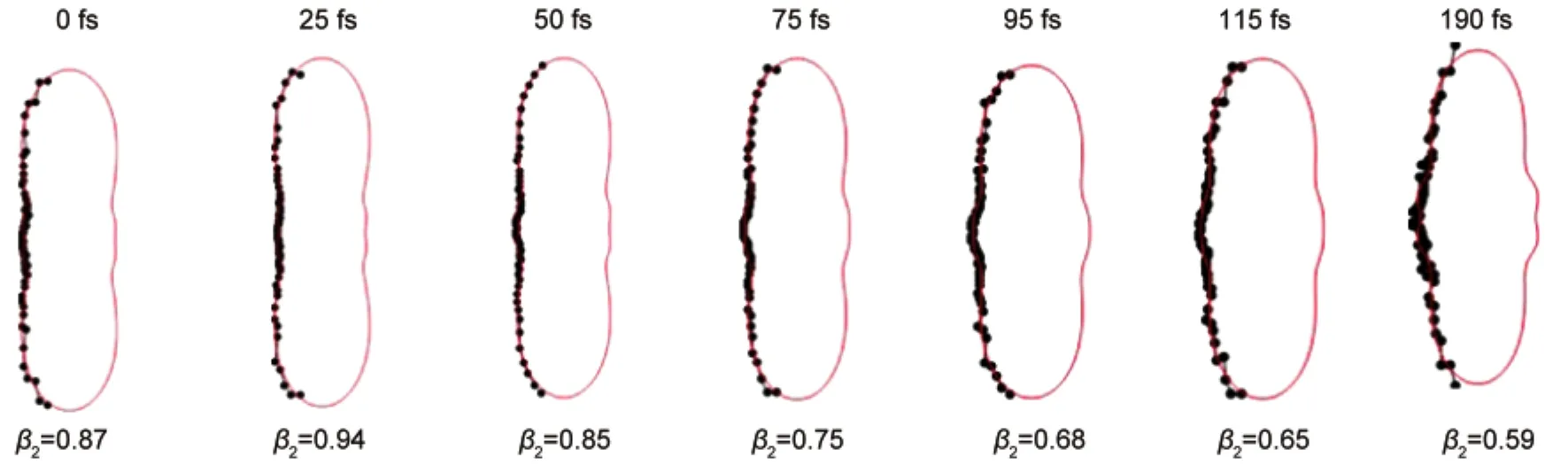
Fig.6 Polar plots of PADs with distributions of PKE of 0.39-1.49 eV observed for different delay timeThe linear polarizations of the pump and probe lasers are aligned vertical in the plane of the figure.
4 Conclusions
We have calculated the transition energy and Franck-Condon factors of the singlet excited states of benzyl chloride molecule.The S4and S2states were decided to have the larger probability to be populated.The ultrafast internal conversion of benzyl chloride was investigated by femtosecond time-resolved photoelectron imaging.By absorption of two photons at 400 nm,benzyl chloride molecule was excited to its S4and S2states.Then the initially excited S4and S2states degenerate in a very short time of 50 fs.This degeneration process can be described by a fast coupling from the S4to S2state and a subsequent internal conversion from the S2to S1state.The following decay of the S1state in a time scale of 910 fs was attributed to another internal conversion to S0state.The time-dependent photoelectron anisotropy parameters in the first 25 fs reflect the coupling between the S4and S2states.
(1) Blanchet,V.;Zgierski,M.Z.;Seideman,T.;Stolow,A.Nature 1999,401,52.doi:10.1038/43410
(2) Schick,C.P.;Carpenter,S.D.;Weber,P.M.J.Phys.Chem.A 1999,103,10470.doi:10.1021/jp992065y
(3) Schmitt,M.;Lochbrunner,S.;Shaffer,J.P.;Larsen,J.J.; Zgierski,M.Z.;Stolow,A.J.Chem.Phys.2001,114,1206.doi: 10.1063/1.1331637
(4) Suzuki,Y.I.;Horio,T.;Fuji,T.;Suzuki,T.J.Chem.Phys.2011, 134,184313.doi:10.1063/1.3586809
(5) Sobolewski,A.L.;Domcke,W.Eur.Phys.J.D 2002,20,369. doi:10.1140/epjd/e2002-00164-5
(6) Suzuki,T.;Wang,L.;Kohguchi,H.J.Chem.Phys.1999,111, 4859.doi:10.1063/1.479822
(7) Wang,L.;Kohguchi,H.;Suzuki,T.Faraday Discuss 1999,113, 37.doi:10.1039/a902866h
(8)Tsubouchi,M.;Whitaker,B.J.;Wang,L.;Kohguchi,H.; Suzuki,T.Phys.Rev.Lett.2001,86,4500.doi:10.1103/ PhysRevLett.86.4500
(9) Song,J.K.;Tsubouchi,M.;Suzuki,T.J.Chem.Phys.2001, 115,8810.doi:10.1063/1.1410974
(10) Stolow,A.Annu.Rev.Phys.Chem.2003,54,89.doi:10.1146/ annurev.physchem.54.011002.103809
(11) Lee,S.H.;Tang,K.C.;Chen,I.C.;Schmitt,M.;Shaffer,J.P.; Schultz,T.;Underwood,J.G.;Zgierski,M.Z.;Stolow,A. J.Phys.Chem.A 2002,106,8979.doi:10.1021/jp021096h
(12) Porter,G.;Windsor,M.W.Nature 1957,180,187.doi:10.1038/ 180187a0
(13)Andre,J.C.;Tournier,A.;Bouchy,M.;Deglise,X.React.Kinet. Catal.Lett.1981,17,433.doi:10.1007/BF02065861
(14) Ichimura,T.;Mori,Y.J.Chem.Phys.1972,57,1677.doi: 10.1063/1.1678454
(15)Ichimura,T.;Mori,Y.;Sumitani,M.;Yoshihara,K.J.Chem. Phys.1984,80,962.doi:10.1063/1.446758
(16) Kawai,A.;Okutsu,T.;Obi,K.Chem.Phys.Lett.1992,198, 637.doi:10.1016/0009-2614(92)85041-8
(17) Nagano,M.;Suzuki,T.;Ichimura,T.;Okutsu,T.;Hiratsuka,H.; Kawauchi,S.J.Phys.Chem.A 2005,109,5825.doi:10.1021/ jp051183k
(18) Matsumoto,R.;Suzuki,T.;Ichimura,T.J.Phys.Chem.A 2005, 109,3331.doi:10.1021/jp044131o
(19)Hiratsuka,H.;Okamoto,T.;Kuroda,S.;Okutsu,T.;Maeoka,H.; Taguchi,M.;Yoshinaga,T.Res.Chem.Intermed.2001,27,137. doi:10.1163/156856701745032
(20) Cristol,S.J.;Bindel,T.H.J.Org.Chem.1980,45,951.doi: 10.1021/jo01294a007
(21) Cristol,S.J.;Bindel,T.H.J.Am.Chem.Soc.1981,103,7287. doi:10.1021/ja00414a041
(22) Cao,Z.Z.;Wei,Z.R.;Hua,L.Q.;Hu,C.J.;Zhang,S.;Zhang, B.ChemPhysChem 2009,10,1299.doi:10.1002/cphc.v10:8
(23) Qin,C.C.;Liu,Y.Z.;Zhang,S.;Wang,Y.M.;Tang,Y.;Zhang, B.Phys.Rev.A 2011,83,033423.doi:10.1103/PhysRevA. 83.033423
(24) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09, RevisionA.02;Gaussian Inc.:Wallingford,CT,2009.
(25)Amma,R.A.;Nair,K.P.R.;Rai,D.K.Appl.Spectrosc.1969, 23,616.doi:10.1366/000370269774380301
(26) Dribinski,V.;Ossadtchi,A.;Mandelshtam,V.A.;Reisler,H. Rev.Sci.Instrum.2002,73,2634.doi:10.1063/1.1482156
(27)Akopyan,M.E.;Vilesov,F.I.;Lopatin,S.N.Khim.Vys.Energ. 1972,6,110.
(28) Yang,C.N.Phys.Rev.1948,74,76.
August 24,2012;Revised:October 11,2012;Published on Web:October 12,2012.
Ultrafast Internal Conversion Dynamics of Benzyl Chloride by Femtosecond Time-Resolved Photoelectron Imaging
DING Zhong-Hua QIU Xue-Jun XU Yan-Qi WANG Yan-Mei*ZHANG Bing
(State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics,Wuhan Institute of Physics and Mathematics, Chinese Academy of Sciences,Wuhan 430071,P.R.China; University of Chinese Academy of Sciences, Beijing 100049,P.R.China)
The ultrafast internal conversion of benzyl chloride(BzCl)was studied with femtosecond time-resolved photoelectron imaging(TRPEI)coupled with time-resolved mass spectroscopy.Time-energy maps of the photoelectron intensity and the angular anisotropy were generated from a series of photoelectron images.Upon absorption of two 400 nm photons,benzyl chloride was excited to the S4and S2states at the same time.The time evolution of the parent ion with different pump-probe delays can be well described by biexponential decay.The fit yielded τ1=50 fs and τ2=910 fs.By analysis of time-resolved photoelectron kinetic energy distributions,it is concluded that the excited S4state has coupled with and decayed to the S2state in a short time scale and then converted to the S1state through ultrafast internal conversion(IC).Within 50 fs,the molecule electronically relaxes into S1through IC and from there,decays to the S0ground state with the relatively slow time constant of 910 fs.The anisotropy parameters of photoelectron angular distributions changed from 0.87 at the delay time of 0 fs to 0.94 at 25 fs and then to 0.59 at 190 fs,which also reflects the coupling from the S4state to the S2state and the following IC to the S1state.
Benzyl chloride;Photoelectron imaging;Internal conversion;Pump-probe; Time-resolved spectroscopy
10.3866/PKU.WHXB201210124
?Corresponding author.Email:meirwang@wipm.ac.cn;Tel:+86-27-87198491.
The project was supported by the National Natural Science Foundation of China(21173256,91121006).
國家自然科學(xué)基金(21173256,91121006)資助項目
O644
猜你喜歡
中學(xué)生數(shù)理化·八年級物理人教版(2023年11期)2023-12-26 07:50:02
井岡教育(2022年2期)2022-10-14 03:11:44
中學(xué)生數(shù)理化(高中版.高考理化)(2022年5期)2022-06-01 06:27:46
中學(xué)生數(shù)理化(高中版.高考理化)(2022年1期)2022-04-26 13:53:42
中學(xué)生數(shù)理化(高中版.高考理化)(2022年3期)2022-04-26 13:41:26
中學(xué)生數(shù)理化(高中版.高考理化)(2022年3期)2022-04-26 13:41:24
課堂內(nèi)外(初中版)(2022年2期)2022-02-28 02:00:30
云南教育·中學(xué)教師(2020年9期)2020-11-16 00:27:58
中學(xué)生數(shù)理化·八年級物理人教版(2019年9期)2019-11-25 07:33:00
中學(xué)生數(shù)理化·八年級物理人教版(2017年9期)2017-12-20 08:11:28
