統(tǒng)計(jì)方法校正O3LYP方法計(jì)算的生成熱
2012-12-11 09:26:28王秀軍
物理化學(xué)學(xué)報(bào) 2012年11期
王秀軍 龍 汨
(華南理工大學(xué)化學(xué)與化工學(xué)院,廣東省燃料電池技術(shù)重點(diǎn)實(shí)驗(yàn)室,廣州510640)
統(tǒng)計(jì)方法校正O3LYP方法計(jì)算的生成熱
王秀軍*龍 汨
(華南理工大學(xué)化學(xué)與化工學(xué)院,廣東省燃料電池技術(shù)重點(diǎn)實(shí)驗(yàn)室,廣州510640)
由于引入各種內(nèi)在近似,密度泛函理論存在固有誤差.本文采用O3LYP/6-311+G(3df,2p)//O3LYP/ 6-31G(d)計(jì)算了220個(gè)中小型有機(jī)分子的生成熱(ΔfH?calc),隨后應(yīng)用神經(jīng)網(wǎng)絡(luò)(ANN)和多元線性回歸(MLR)方法對(duì)ΔfH?calc進(jìn)行校正.采用計(jì)算得到的生成熱、零點(diǎn)能、分子中原子總數(shù)、氫原子個(gè)數(shù)、雙中心成鍵電子數(shù)、雙中心反鍵電子數(shù)、單中心價(jià)層孤對(duì)電子數(shù)、單中心內(nèi)層電子數(shù)作為ANN和MLR的描述符.以180個(gè)分子作為訓(xùn)練集構(gòu)造ANN或MLR模型,并對(duì)40個(gè)獨(dú)立測(cè)試集分子的ΔfH?calc進(jìn)行了預(yù)測(cè).結(jié)果表明:經(jīng)過ANN和MLR校正后,訓(xùn)練集分子生成熱的理論計(jì)算值和實(shí)驗(yàn)值間的均方根偏差(RMSD)從24.7 kJ·mol-1分別降低到11.8、13.0 kJ· mol-1;獨(dú)立測(cè)試集分子的RMSD從21.3 kJ·mol-1分別降低到10.4、12.1 kJ·mol-1.因此ANN模型的擬合和預(yù)測(cè)能力要明顯優(yōu)于MLR模型.
O3LYP;神經(jīng)網(wǎng)絡(luò);多元線性回歸;生成熱;均方根偏差
1 引言
分子的能量是分子最基本的物理量.在實(shí)際研究中無法測(cè)定體系精確的能量,因此我們更關(guān)心那些能夠表征體系能量的熱力學(xué)函數(shù),其中生成熱(ΔfH?)就是一個(gè)最基本的表征化合物在標(biāo)準(zhǔn)狀態(tài)下穩(wěn)定性的熱力學(xué)量,對(duì)判斷有機(jī)反應(yīng)的方向和限度也十分重要,并且實(shí)驗(yàn)上可以測(cè)得其精確值,1-3因此生成熱的應(yīng)用十分廣泛.但是實(shí)驗(yàn)上要精確測(cè)定一個(gè)化合物的生成熱也是很困難的,不僅需要一套合適的裝置,還需要花費(fèi)大量的時(shí)間,并且還需要測(cè)定者有很豐富的測(cè)定經(jīng)驗(yàn),而且不同的計(jì)算方法間也會(huì)有偏差,4-6有時(shí)新測(cè)定的結(jié)果還會(huì)否定舊的實(shí)驗(yàn)結(jié)果,7因此近年測(cè)定新的化合物生成熱數(shù)據(jù)的報(bào)導(dǎo)很少,于是發(fā)展一個(gè)簡(jiǎn)單的并能準(zhǔn)確預(yù)測(cè)生成熱的計(jì)算模型是非常必要的.
隨著計(jì)算機(jī)技術(shù)的飛速發(fā)展,科學(xué)工作者對(duì)計(jì)算化學(xué)越來越依賴,從而計(jì)算化學(xué)已成為對(duì)實(shí)驗(yàn)結(jié)果的解釋和預(yù)測(cè)不可缺少的工具.8與經(jīng)典從頭算方法相比,密度泛函理論(DFT)具有計(jì)算花費(fèi)少,而且可以計(jì)算更大的分子,它在計(jì)算量子化學(xué)領(lǐng)域廣泛應(yīng)用,已經(jīng)成為化學(xué)、凝聚態(tài)物理、材料科學(xué)和分子生物學(xué)中重要的研究工具,但是DFT計(jì)算仍然存在一些計(jì)算偏差.例如目前最流行的雜化密度泛函B3LYP計(jì)算存在一些缺陷:隨著系統(tǒng)增大偏差增大;9,10它所計(jì)算鍵的離解焓偏低;11,12得到不準(zhǔn)確的異構(gòu)體能量差別;12低估了反應(yīng)能壘;13,14不能準(zhǔn)確計(jì)算分子間的范德華作用力.15Cohen和Handy16在B3LYP的基礎(chǔ)上發(fā)展了一種新的泛函—O3LYP,他們引入一個(gè)交換泛函OPTX.近幾年的研究發(fā)現(xiàn)O3LYP的計(jì)算結(jié)果要優(yōu)于B3LYP.17-23
雖然Gn(n=1-4)方法在計(jì)算小分子生成熱方面成效非常顯著,9,24-29但是它們的計(jì)算花費(fèi)不經(jīng)濟(jì),科學(xué)家一般不利用此類方法計(jì)算大分子的生成熱.另一方面,由于DFT計(jì)算的一些固有偏差,其局限性也越來越突出,尤其是在計(jì)算大分子體系時(shí)更是如此.因此,一些學(xué)者提出了一些校正模型對(duì)DFT計(jì)算結(jié)果進(jìn)行校正,并取得了令人滿意的結(jié)果. Chen等30,31首次采用ANN校正了密度泛函理論的計(jì)算結(jié)果,生成熱的計(jì)算值與實(shí)驗(yàn)值之間的均方根偏差從89.5 kJ·mol-1(或50.2 kJ·mol-1)降為13.0 kJ· mol-1(或13.8 kJ·mol-1).Fan等32提出了用自然鍵軌道(NBO)分析并運(yùn)用神經(jīng)網(wǎng)絡(luò)校正350個(gè)分子的計(jì)算生成熱,減少了350個(gè)分子的生成熱的計(jì)算偏差,隨后他們運(yùn)用多元線性回歸算法校正DFT計(jì)算的生成熱,也成功地減小了DFT的計(jì)算偏差.33Li等34利用支持向量機(jī)模型也成功地校正了DFT計(jì)算的生成熱.這說明神經(jīng)網(wǎng)絡(luò),多元線性回歸,支持向量機(jī)等統(tǒng)計(jì)方法是適合于校正DFT計(jì)算結(jié)果的. Chen35及本課題組36也采用神經(jīng)網(wǎng)絡(luò)模型通過調(diào)節(jié)B3LYP雜化密度泛函中三個(gè)參數(shù)來減少密度泛函理論計(jì)算的分子熱力學(xué)性質(zhì)數(shù)值同實(shí)驗(yàn)值的偏差,并取得了成功.本文利用ANN和MLR的方法來校正O3LYP計(jì)算的220個(gè)有機(jī)分子的生成熱,并對(duì)這兩種校正方法的計(jì)算結(jié)果進(jìn)行比較.
2 方法理論
2.1 計(jì)算方法
本文選用的是包括小型(重原子數(shù)目<6)和中型(重原子數(shù)目≥6)的220個(gè)有機(jī)分子,所有的分子都是由H、C、N、O、F、S、Cl、Br這幾種原子組成,每個(gè)分子中含有1-12個(gè)C原子,這些分子的生成熱實(shí)驗(yàn)值都是從實(shí)驗(yàn)手冊(cè)中獲得.1-3所有分子在這三本手冊(cè)中的ΔfH?的實(shí)驗(yàn)值偏差都小于4 kJ·mol-1.30在上述分子中,選取文獻(xiàn)30中的180個(gè)分子作為訓(xùn)練集用于構(gòu)造ANN或MLR模型,利用交叉驗(yàn)證的方法來檢驗(yàn)ANN和MLR的校正結(jié)果.本文首先用O3LYP/ 6-31G(d)方法優(yōu)化所有分子的結(jié)構(gòu),然后用O3LYP/ 6-311+G(3df,2p)方法計(jì)算其生成熱,作為訓(xùn)練集的180個(gè)分子和測(cè)試集的40個(gè)分子計(jì)算結(jié)果與實(shí)驗(yàn)值的RMS偏差分別是24.7、21.3 kJ·mol-1.對(duì)所有分子進(jìn)行了NBO分析.NBO分析得到的分子的結(jié)構(gòu)參數(shù)用于構(gòu)造ANN(MLR)模型的描述符(自變量).所有O3LYP計(jì)算均采用Gaussian 09程序包.37
在構(gòu)造統(tǒng)計(jì)校正模型時(shí),最重要的一點(diǎn)就是關(guān)于分子的物理描述符的選擇,因?yàn)檫@些描述符被用作神經(jīng)網(wǎng)絡(luò)的輸入層和多元線性回歸的自變量.首先我們選擇O3LYP計(jì)算的生成熱()作為統(tǒng)計(jì)模型的第一個(gè)描述符.Chen等30在利用神經(jīng)網(wǎng)絡(luò)改進(jìn)B3LYP計(jì)算的時(shí),他們發(fā)現(xiàn)對(duì)神經(jīng)網(wǎng)絡(luò)的校正結(jié)果非常顯著,因此可以作為第一個(gè)描述符.另外Gn(n=1-4)在計(jì)算中小型分子生成熱時(shí)效果顯著,這是因?yàn)樵贕n方法中引入了一個(gè)高階校正(HLC)項(xiàng)來抵消一系列計(jì)算后剩余能量偏差,26而HLC主要采用了分子和原子中的價(jià)層孤對(duì)電子和未成對(duì)電子對(duì)其能量進(jìn)行最后擬合得到.Gn方法認(rèn)為大部分的相關(guān)能是由占據(jù)在同一個(gè)分子軌道上的電子之間的相互作用引起的,因此電子越多,它們的相關(guān)能就越大,且成對(duì)電子對(duì)相關(guān)能的貢獻(xiàn)和未成對(duì)電子是不一樣的.而且G4對(duì)原子和分子采用不同的參數(shù),說明電子在分子環(huán)境和原子環(huán)境中,它們對(duì)相關(guān)能的貢獻(xiàn)也不一樣.鑒于以上分析,本文也采用NBO計(jì)算分子的雙中心成鍵電子數(shù)(BD)、雙中心反鍵電子數(shù)(BD*)、單中心價(jià)層孤對(duì)電子數(shù)(LP)、單中心的內(nèi)層電子數(shù)(CR)這四個(gè)參數(shù)作為描述符.考慮到分子的大小對(duì)的計(jì)算偏差有很大的影響,其偏差隨著原子數(shù)的增加而遞增,因而把原子總數(shù)(NT)和氫原子個(gè)數(shù)(NH)作為描述符.分子的零點(diǎn)能(ZPE)是計(jì)算的一個(gè)重要參數(shù),因此將ZPE作為另一個(gè)重要的描述符.30
2.2 多元線性回歸模型
采用SPSS11.01統(tǒng)計(jì)軟件分析180個(gè)分子訓(xùn)練集的上述描述符參數(shù)與生成熱實(shí)驗(yàn)值的關(guān)聯(lián)度,并構(gòu)建實(shí)驗(yàn)值的多元線性回歸方程如下:
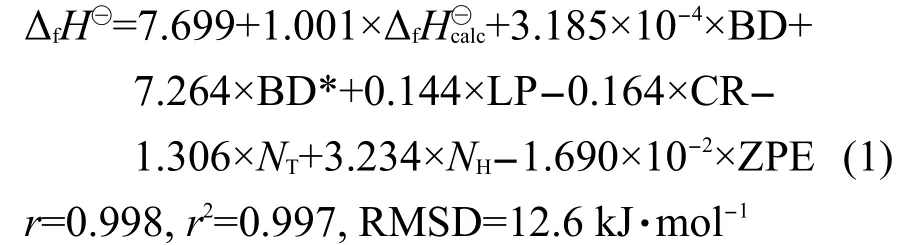
其中,ΔfH?為應(yīng)用MLR方程校正后的生成熱值,r為相關(guān)系數(shù),RMSD為均方根偏差.
2.3 神經(jīng)網(wǎng)絡(luò)模型
神經(jīng)網(wǎng)絡(luò)是一個(gè)由許多簡(jiǎn)單的并行工作的處理單元組成的系統(tǒng),其功能取決于網(wǎng)絡(luò)的結(jié)構(gòu)、連接強(qiáng)度以及各單元的處理方式.所采用的神經(jīng)網(wǎng)絡(luò)模型包含有輸入層,隱藏層和輸出層三層.本文中的神經(jīng)網(wǎng)絡(luò)結(jié)構(gòu)見圖1,其中輸入層(X1,X2,X3,…)是由8個(gè)描述符和一個(gè)偏置量(bias)組成,隱藏層包含有3個(gè)隱藏神經(jīng)元(y1,y2,y3),一個(gè)輸出層是輸出校正后的分子生成熱.{Wxij}和{Wyj}表示突觸權(quán)重,其中{Wxij}是連接輸入神經(jīng)元和隱藏神經(jīng)元的突觸權(quán)重,{Wyj}是連接隱藏神經(jīng)元和輸出神經(jīng)元的突觸權(quán)重.在神經(jīng)網(wǎng)絡(luò)模型中所用的轉(zhuǎn)換函數(shù)是sigmoid函數(shù).輸出層Z與輸入層Xi的關(guān)系由公式(2)和(3)描述:
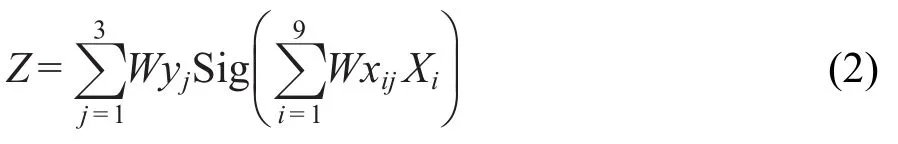
其中,

這里Z為經(jīng)ANN校正后的生成熱值,Xi對(duì)應(yīng)圖1中給出的8個(gè)描述符和一個(gè)偏置量,Sig(v)為sigmoid函數(shù).
2.4 模型的驗(yàn)證
本文采用六重交叉驗(yàn)證的方法來驗(yàn)證MLR和ANN模型的校正結(jié)果,即將180個(gè)分子分成兩組,其中一組共有150個(gè)分子作為訓(xùn)練集,另一組30個(gè)分子作為測(cè)試集,這個(gè)過程循環(huán)重復(fù)了6次.同時(shí)計(jì)算評(píng)價(jià)模型擬合能力的標(biāo)準(zhǔn)回歸系數(shù)(q2)和RMSD. Tropsha等38將q2定義為:
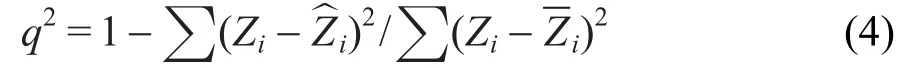
其中,Zi和分別表示物種的實(shí)驗(yàn)值和預(yù)測(cè)值,Zi表示物種訓(xùn)練值的平均值,因此本文采用的計(jì)算公式如下:
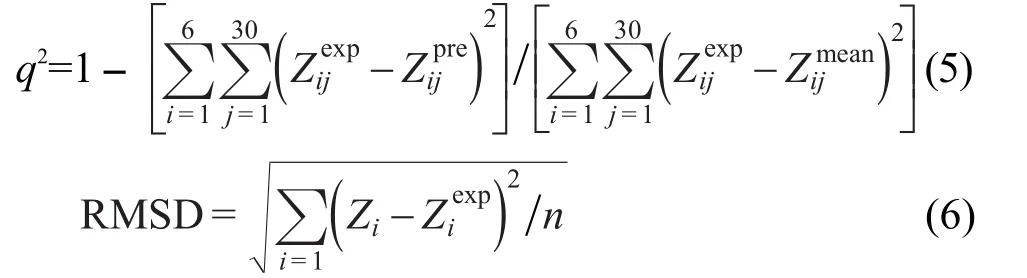
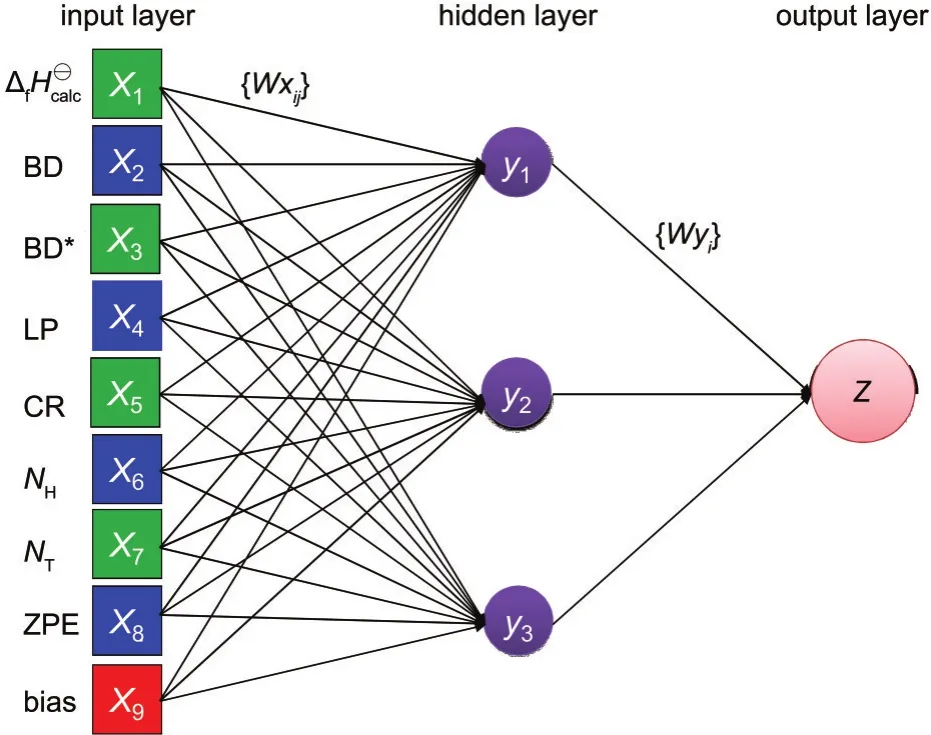
圖1 神經(jīng)網(wǎng)絡(luò)的結(jié)構(gòu)Fig.1 Structure of the neural networkXi(i=1-9)are ΔfH?calc,the total quantities of atoms(NT),hydrogen atoms(NH),2-center bond(BD),2-center antibond(BD*),1-center valence lone pair(LP),and 1-center core pair(CR),the zero point energy(ZPE)and bias,respectively.yj(j=1-3)are the number of hidden neuron.{Wxij}are the synaptic weights connecting the input layer(Xi)and the hidden neurons(yj)and{Wyj}connect the hidden neurons and the output(Z).Z is the ΔfH?corrected byANN.
3 結(jié)果與討論
3.1 MLR校正生成熱
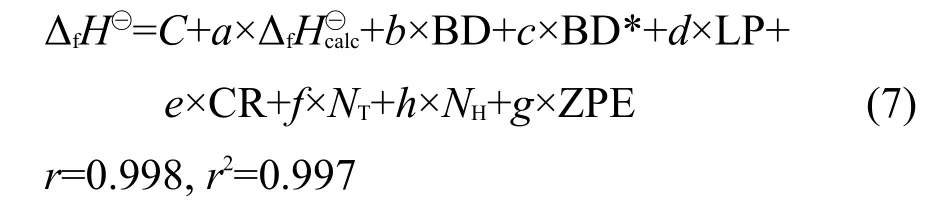
其中,系數(shù)a-g對(duì)每次回歸分析都有不同的數(shù)值,與方程(1)相比,經(jīng)過交叉驗(yàn)證后得到的r與r2幾乎不變,MLR的結(jié)果分析見表1.由表1可知,MLR經(jīng)交叉驗(yàn)證后的q2值是-0.53,訓(xùn)練集的平均RMSD是12.5 kJ·mol-1,但是測(cè)試集的平均RMSD高達(dá)15.1 kJ·mol-1,總體RMSD是13.0 kJ·mol-1.Cramer等39曾指出q2為負(fù)值是因?yàn)闇y(cè)試集的平均偏差要大于訓(xùn)練集的平均偏差,說明模型預(yù)測(cè)能力較差.Golbraikh和Tropsha40曾提出統(tǒng)計(jì)參數(shù)q2>0.5,r2>0.6的模型有效,同時(shí)Tropsha等也認(rèn)為模型的預(yù)測(cè)能力比擬合能力更為重要,即q2的大小更為關(guān)鍵.由上述實(shí)驗(yàn)結(jié)果可知,利用MLR來預(yù)測(cè)中小型分子的生成熱是不可靠的.MLR雖然在訓(xùn)練集中可以降低生成熱的RMSD,但是其模型的預(yù)測(cè)能力較差.
3.2 神經(jīng)網(wǎng)絡(luò)校正生成熱
圖2是O3LYP計(jì)算結(jié)果、MLR校正結(jié)果(O3LYP-MLR)、ANN校正結(jié)果(O3LYP-ANN)和實(shí)驗(yàn)值的對(duì)比以及偏差直方圖.由圖2(a,b)可知,與實(shí)驗(yàn)值相比,原始的O3LYP計(jì)算結(jié)果存在很大的系統(tǒng)偏差,偏差在±4 kJ·mol-1范圍內(nèi)的分子個(gè)數(shù)只有24個(gè),經(jīng)過神經(jīng)網(wǎng)絡(luò)校正后(見圖2(e,f)),生成熱的校正結(jié)果明顯更加接近于實(shí)驗(yàn)值,偏差分布非常集中,所有分子的偏差范圍都集中在±32 kJ·mol-1的范圍內(nèi),而且偏差在±4 kJ·mol-1范圍內(nèi)的分子個(gè)數(shù)有63個(gè),明顯好于O3LYP的計(jì)算結(jié)果.MLR盡管也能在一定程度上對(duì)O3LYP的計(jì)算結(jié)果進(jìn)行校正,但其模型預(yù)測(cè)能力較差.
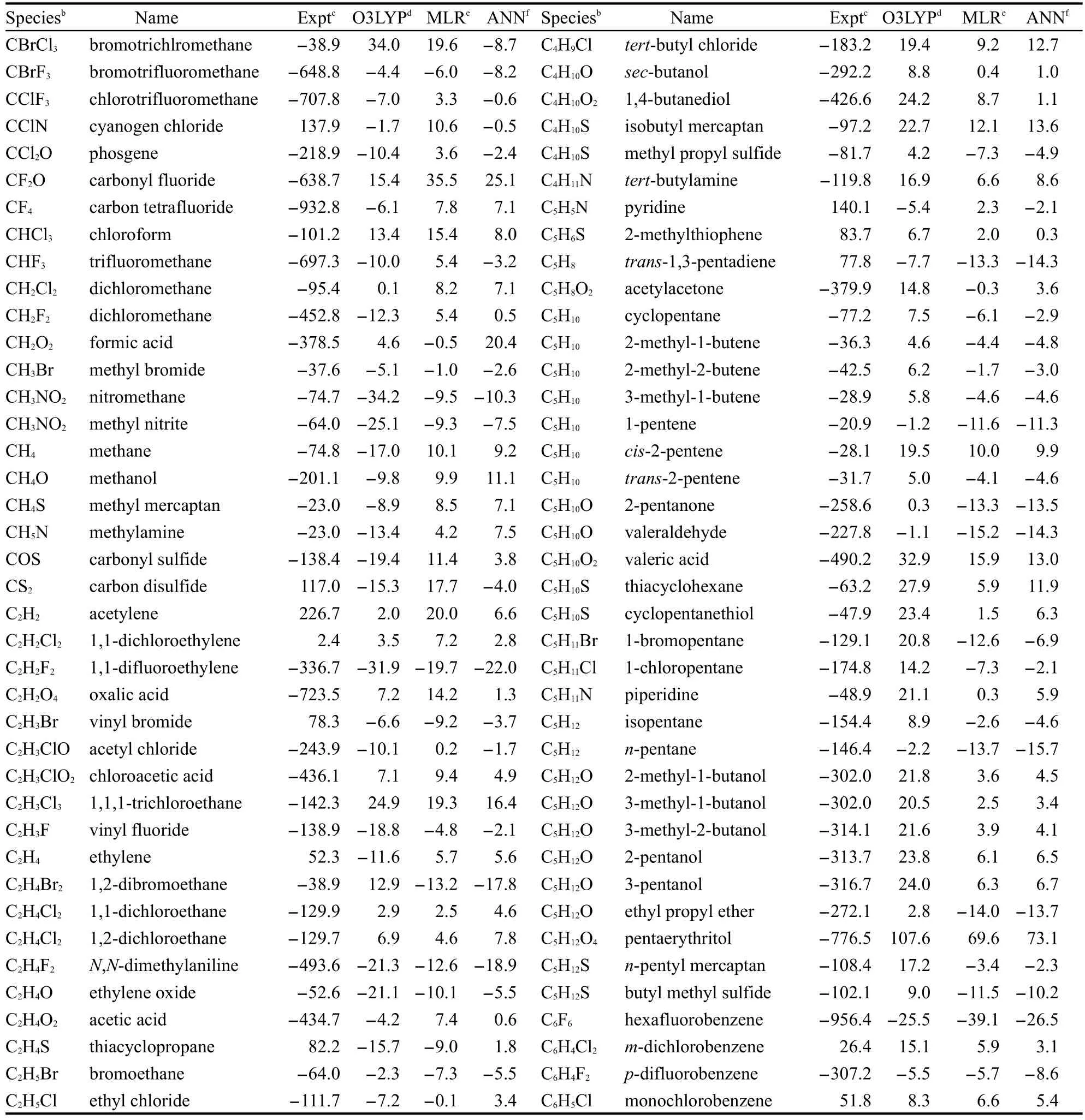
表3 ΔfH?的實(shí)驗(yàn)值和不同方法的計(jì)算值和實(shí)驗(yàn)值間的偏差aTable 3 Experimental ΔfH?and the deviation between the calculated and experimental values with different methodsa
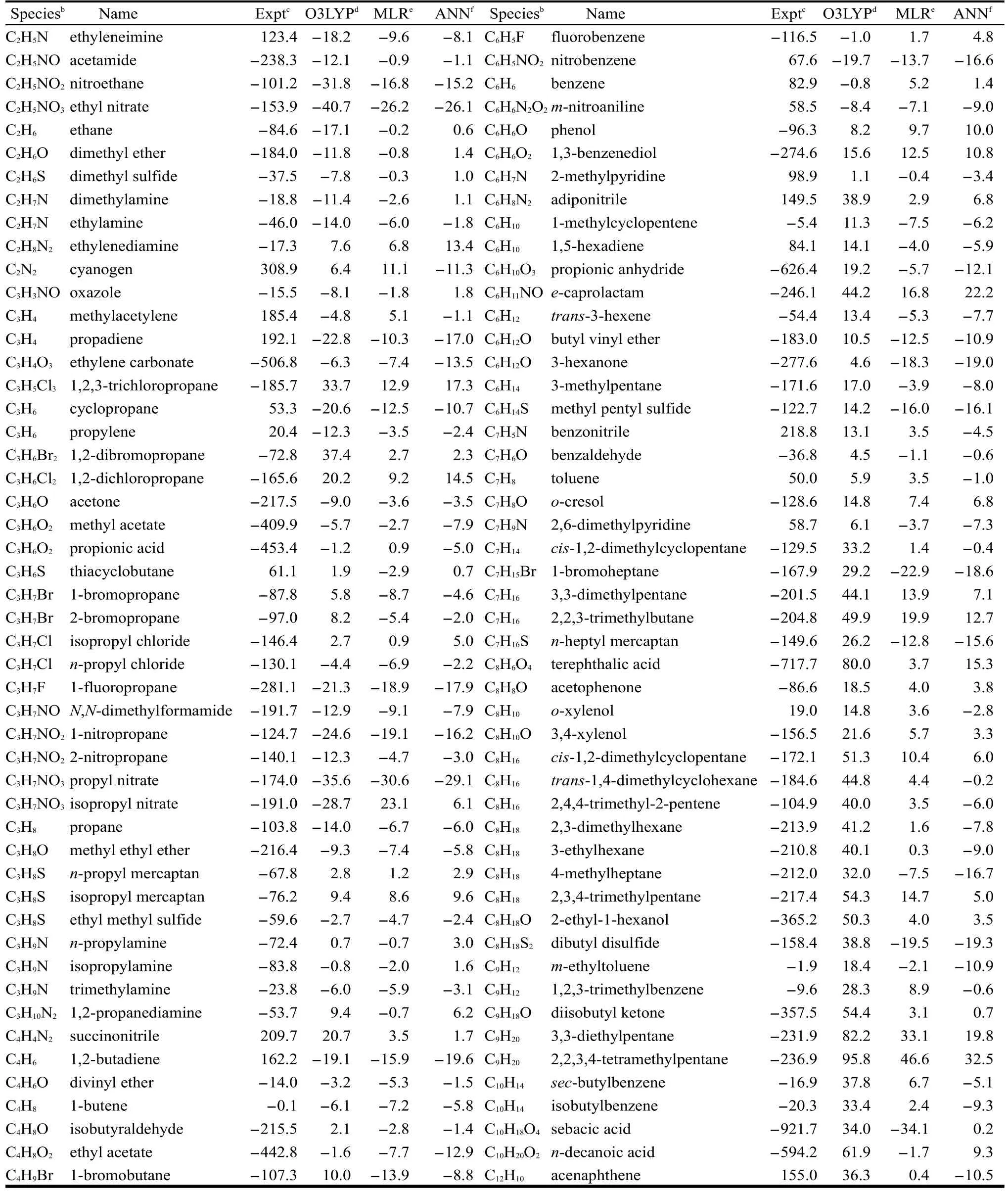
continued Table 3

表4 測(cè)試集ΔfH?的實(shí)驗(yàn)值和不同方法的計(jì)算值和實(shí)驗(yàn)值間的偏差aTable 4 Experimental ΔfH?of testing set and the deviation between the calculated and experimental values with different methodsa
180個(gè)訓(xùn)練集分子經(jīng)ANN校正后的結(jié)果列于表3.為了對(duì)比,表3中也列出了O3LYP的計(jì)算值與實(shí)驗(yàn)值間的偏差,MLR得到的方程(1)擬合的計(jì)算值與實(shí)驗(yàn)值間的偏差及全部分子的ΔfH?實(shí)驗(yàn)值.
綜上所述,不管是模型的擬合還是模型的預(yù)測(cè),利用ANN校正生成熱的結(jié)果要明顯優(yōu)于MLR模型.原因可能是ANN模型采用了Sigmod函數(shù)來校正理論生成熱,而MLR模型不是利用非線性函數(shù).在ANN模型里,每個(gè)輸入的參數(shù)都與隱藏層的神經(jīng)元有一定的關(guān)系,而本文所采用的ANN模型的輸入層中包含一個(gè)偏置量,而且隱藏層包含三個(gè)神經(jīng)元,因此ANN在擬合的過程中產(chǎn)生的變量數(shù)目要比MLR多,而在MLR方法中,變量的數(shù)目與輸入的參數(shù)數(shù)目相同.
3.3 獨(dú)立測(cè)試集的預(yù)測(cè)結(jié)果
為了驗(yàn)證MLR和ANN模型的預(yù)測(cè)能力,我們?nèi)〕?0個(gè)分子建立了一個(gè)獨(dú)立測(cè)試集,經(jīng)過MLR和ANN模型校正后,測(cè)試集分子生成熱的RMSD從21.3 kJ·mol-1分別降低到12.1、10.4 kJ·mol-1.因此,ANN模型的預(yù)測(cè)能力要優(yōu)于MLR模型.表4列出了獨(dú)立測(cè)試集分子經(jīng)MLR和ANN校正后的結(jié)果,同時(shí)也列出了生成熱的O3LYP計(jì)算值與實(shí)驗(yàn)值間的偏差.
4 結(jié)論
采用O3LYP/6-311+G(3df,2p)//O3LYP/6-31G(d)計(jì)算220個(gè)有機(jī)分子的生成熱,然后采用ΔfH?calc、雙中心成鍵電子數(shù)、雙中心反鍵電子數(shù)、單中心價(jià)層孤對(duì)電子數(shù)、單中心的內(nèi)層電子數(shù)、原子總數(shù)和氫原子個(gè)數(shù)、分子的零點(diǎn)能作為描述符參數(shù),運(yùn)用神經(jīng)網(wǎng)絡(luò)方法和多元線性回歸方法對(duì)生成熱進(jìn)行校正.研究結(jié)果表明:經(jīng)過神經(jīng)網(wǎng)絡(luò)校正后,ΔfH?的理論計(jì)算值與實(shí)驗(yàn)值間的RMSD從24.7 kJ·mol-1降低到11.8 kJ·mol-1,ANN模型的標(biāo)準(zhǔn)回歸系數(shù)是0.96, MLR雖然在一定程度上降低了生成熱的RMSD,但是它的標(biāo)準(zhǔn)回歸系數(shù)很小,說明MLR的預(yù)測(cè)能力很差.采用ANN和MLR模型校正40個(gè)獨(dú)立測(cè)試集分子的生成熱,結(jié)果表明其RMSD從21.3 kJ·mol-1分別降低到10.4、12.1 kJ·mol-1.因此ANN模型的擬合和預(yù)測(cè)能力要明顯優(yōu)于MLR.在利用統(tǒng)計(jì)方法校正能量時(shí),描述符的選取是問題的關(guān)鍵,因此有必要選擇合適的描述符,從而進(jìn)一步改善統(tǒng)計(jì)方法對(duì)理論計(jì)算獲得的能量的校正結(jié)果.
(1) Pedley,J.B.;Naylor,R.D.;Kirby,S.P.Thermochemical Data of Organic Compounds;Chapman and Hall:New York,1986.
(2)Yaws,C.L.Chemical Properties Handbook;McGraw-Hill: New York,1999.
(3) Lide,D.R.CRC Handbook of Chemistry and Physics,3rd. electronic ed.;BocaRaton:FL,2000.
(4) Wu,J.;Xu,X.J.Chem.Phys.2007,127,214105.doi:10.1063/ 1.2800018
(5) Curtiss,L.A.;Raghavachari,K.;Redfern,P.C.;Pople,J.A. Chem.Phys.Lett.1997,270,419.doi:10.1016/S0009-2614(97) 00399-0
(6) Schmitz,L.R.;Chen,K.H.;Labanowski,J.;Allinger,N.L. J.Phys.Org.Chem.2001,14,90.doi:10.1002/1099-1395 (200102)14:2<90::AID-POC330>3.0.CO;2-O
(7) Curtiss,L.A.;Raghavachari,K.;Trucks,G.W.;Pople,J.A. J.Chem.Phys.1991,94,7221.doi:10.1063/1.460205
(8) Lado-Tourino,I.;Tsobnang,F.Comp.Mater.Sci.1998,11,181. doi:10.1016/S0927-0256(98)80004-9
(9) Curtiss,L.A.;Raghavachari,K.;Redfern,P.C.;Pople,J.A. J.Chem.Phys.2000,112,7374.doi:10.1063/1.481336
(10) Wodrich,M.D.;Corminboeuf,C.;Schleyer,P.v.R.Org.Lett. 2006,8,3631.doi:10.1021/ol061016i
(11) Check,C.E.;Gilbert,T.M.J.Org.Chem.2005,70,9828.doi: 10.1021/jo051545k
(12) Izgorodina,E.I.;Coote,M.L.;Radom,L.J.Phys.Chem.A 2005,109,7558.doi:10.1021/jp052021r
(13) Schreiner,P.R.;Fokin,A.A.;Pascal,R.A.,Jr.;de Meijere,A. Org.Lett.2006,8,3635.doi:10.1021/ol0610486
(14)Zhao,Y.;Gonzalez-Garcia,N.;Truhlar,D.G.J.Phys.Chem.A 2005,109,2012.doi:10.1021/jp045141s
(15) Zhang,I.Y.;Luo,Y.;Xu,X.J.Chem.Phys.2010,132,194105. doi:10.1063/1.3424845
(16) Cohen,A.J.;Handy,N.C.Mol.Phys.2001,99,607.doi: 10.1080/00268970010023435
(17)Yang,K.;Peverati,R.;Truhlar,D.G.;Valero,R.J.Chem.Phys. 2011,135,044118.doi:10.1063/1.3607312
(18) Heerdt,G.;Morgon,N.H.Quimica Nova 2011,34,868.doi: 10.1590/S0100-40422011000500024
(19) Bochevarov,A.D.;Friesner,R.A.;Lippard,S.J.J.Chem. Theory Comput.2010,6,3735.doi:10.1021/ct100398m
(20)Dobado,J.A.;Gomez-Tamayo,J.C.;Calvo-Flores,F.G.; Martinez-Garcia,H.;Cardona,W.;Weiss-Lopez,B.;Ramirez-Rodriguez,O.;Pessoa-Mahana,H.;Araya-Maturana,R.Magn. Reson.Chem.2011,49,358.doi:10.1002/mrc.2745
(21) Qian,Z.S.;Feng,H.;He,L.N.;Yang,W.J.;Bi,S.P.J.Phys. Chem.A 2009,113,5138.doi:10.1021/jp810632f
(22) Strassner,T.;Taige,M.A.J.Chem.Theory Comput.2005,1, 848.doi:10.1021/ct049846+
(23) Baker,J.;Pulay,P.J.Comput.Chem.2003,24,1184.doi: 10.1002/jcc.10280
(24) Curtiss,L.A.;Jones,C.;Trucks,G.W.;Raghavachari,K.; Pople,J.A.J.Chem.Phys.1990,93,2537.doi:10.1063/ 1.458892
(25) Curtiss,L.A.;Raghavachari,K.;Redfern,P.C.;Pople,J.A. J.Chem.Phys.1997,106,1063.doi:10.1063/1.473182
(26) Curtiss,L.A.;Raghavachari,K.;Redfern,P.C.;Rassolov,V.; Pople,J.A.J.Chem.Phys.1998,109,7764.doi:10.1063/ 1.477422
(27) Curtiss,L.A.;Raghavachari,K.;Redfern,P.C.;Pople,J.A. J.Chem.Phys.2000,112,7374.doi:10.1063/1.481336
(28) Curtiss,L.A.;Redfern,P.C.;Raghavachari,K.Chem.Phys. Lett.2010,499,168.doi:10.1016/j.cplett.2010.09.012
(29) Dorofeeva,O.V.;Kolesnikova,I.N.;Marochkin,I.I.;Ryzhova, O.N.J.Struct.Chem.2011,22,1303.doi:10.1007/s11224-011-9827-7
(30)Hu,L.H.;Wang,X.J.;Wong,L.H.;Chen,G.H.J.Chem. Phys.2003,119,11501.doi:10.1063/1.1630951
(31)Wang,X.J.;Wong,L.H.;Hu,L.H.;Chan,C.Y.;Su,Z.M.; Chen,G.H.J.Phys.Chem.A 2004,108,8514.doi:10.1021/ jp047263q
(32)Duan,X.M.;Li,Z.H.;Song,G.L.;Wang,W.N.;Chen,G.H.; Fan,K.N.Chem.Phys.Lett.2005,410,125.doi:10.1016/ j.cplett.2005.05.046
(33)Duan,X.M.;Song,G.L.;Li,Z.H.;Wang,X.J.;Chen,G.H.; Fan,K.N.J.Chem.Phys.2004,121,7086.doi:10.1063/ 1.1786582
(34) Yan,G.K.;Li,J.J.;Li,B.R.;Hu,J.;Guo,W.P.J.Theor. Comput.Chem.2007,6,495.doi:10.1142/ S0219633607003118
(35) Zheng,X.;Hu,L.;Wang,X.;Chen,G.Chem.Phys.Lett.2004, 390,186.doi:10.1016/j.cplett.2004.04.020
(36) Zhang,J.H.;Wang,X.J.Acta Phys.-Chim.Sin.2010,26, 188. [張家虎,王秀軍.物理化學(xué)學(xué)報(bào),2010,26,188.] doi:10.3866/PKU.WHXB20100116
(37) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09, Revision B.01;Gaussian Inc.:Wallingford,CT,2010.
(38) Zheng,W.F.;Tropsha,A.J.Chem.Inf.Comput.Sci.2000,40, 185.doi:10.1021/ci980033m
(39) Cramer,R.D.,III;Patterson,D.E.;Bunce,J.D.J.Am.Chem. Soc.1988,110,5959.doi:10.1021/ja00226a005
(40) Golbraikh,A.;Tropsha,A.J.Chem.Inf.Comput.Sci.2003,43, 144.doi:10.1021/ci025516b
April 9,2012;Revised:July 16,2012;Published on Web:July 17,2012.
Statistical Correction of Heat of Formation Calculated by the O3LYP Method
WANG Xiu-Jun*LONG Mi
(Key Laboratory of Fuel Cell Techology of Guangdong,College of Chemistry and Chemical Engineering, South China University of Technology,Guangzhou 510640,P.R.China)
The results of density functional theory calculations are known to contain inherent numerical errors caused by various intrinsic approximations.In this paper,O3LYP/6-311+G(3df,2p)//O3LYP/6-31G(d) calculations were used to derive the heats of formation(ΔfH?calc)of 220 small to medium-sized organic molecules,followed by the application of artificial neural network(ANN)and multiple linear regression (MLR)analyses to correct the values.The physical descriptors chosen were ΔfH?calcand zero point energy as well as the total quantities of atoms,hydrogen atoms,2-center bonds,2-center antibonds,1-center valence lone pairs and 1-center core pairs.The ANN and MLR systems were initially constructed using a 180 training set.The trained ANN and MLR systems were subsequently used to predict values of ΔfH?calcfor a 40 individual testing set.The results demonstrated that the root mean square(RMS)deviations between the calculated and experimental ΔfH?values in the training set were reduced from 24.7 to 11.8 and 13.0 kJ· mol-1after ANN and MLR corrections,respectively.For the individual testing set,the deviations(RMSD) were reduced from 21.3 to 10.4 and 12.1 kJ·mol-1,respectively.Based on these results,it can be concluded thatANN exhibits superior fitting and predictive abilities compared with MLR.
O3LYP;Neural network;Multiple linear regression;Heat of formation;Root mean square deviation
10.3866/PKU.WHXB201207172
?Corresponding author.Email:xjwangcn@scut.edu.cn;Tel:+86-20-87112943.
The project was supported by the National Natural Science Foundation of China(20975040)and Natural Science Foundation of Guangdong Province,China(10351064101000000).
國(guó)家自然科學(xué)基金(20975040)及廣東省自然科學(xué)基金(10351064101000000)資助項(xiàng)目
O641
猜你喜歡
童話王國(guó)·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2020年10期)2020-11-26 08:24:50
數(shù)學(xué)物理學(xué)報(bào)(2020年2期)2020-06-02 11:29:24
小哥白尼(趣味科學(xué))(2019年6期)2019-10-10 01:01:50
光學(xué)精密工程(2016年6期)2016-11-07 09:07:19
發(fā)明與創(chuàng)新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34