云母表面吸附烷基伯胺對其疏水性的影響
2012-12-21 06:33:18劉夠生于建國
物理化學學報 2012年1期
劉 臻 劉夠生 于建國
(華東理工大學化學工程聯合國家重點實驗室,上海200237)
云母表面吸附烷基伯胺對其疏水性的影響
劉 臻 劉夠生*于建國
(華東理工大學化學工程聯合國家重點實驗室,上海200237)
礦物表面的疏水性受吸附在其表面的表面活性劑的影響,進而影響礦物的浮選行為.本文通過表面接觸角測量、原子力顯微鏡(AFM)觀測以及密度泛函理論(DFT)和分子動力學(MD)模擬計算,研究了吸附在云母表面的烷基伯胺的鏈長對其疏水性的影響.通過比較氧密度和氫鍵數量分布,發現每個水分子在碳氫鏈尾端和水相接觸的界面上相對于在體相中形成氫鍵的能力有所降低,而吸附烷基伯胺的云母由親水性轉化為疏水性.研究結果還表明,在單分子層吸附狀態下,吸附十八胺的云母的疏水性比吸附十二胺的云母的疏水性要強,且由于十八胺的臨界半膠束濃度(HMC)要遠低于十二胺,十八胺更易在云母表面形成多層吸附,證明烷基伯胺的碳鏈越長,其對云母表面疏水性改善的能力越強.實驗結果與理論計算結果吻合良好.
云母;吸附;疏水性;原子力顯微鏡;分子動力學模擬
1 引言
表面活性劑在固體表面的吸附和聚集是膠體與界面化學中一個十分重要的領域,研究手段非常豐富,包括原子力顯微鏡(AFM),1-4傅里葉變換紅外(FTIR)光譜,5和頻振動光譜(SFG),6X射線光電子能譜(XPS)5等.另外,分子模擬方法在近20年來也越來越多地應用在這一領域,6-11包括從量子力學,分子動力學,到介觀動力學等方法,跨越了從納米到微米,從飛秒到毫秒的時空范圍.
我國江西宜春鉭鈮礦采用椰油伯胺從鉭鈮尾礦中浮選鋰云母,椰油伯胺是一種包含8-18個碳原子的烷基伯胺的混合胺,經分析其中主要包含十二胺(DDA)和十八胺(ODA).烷基伯胺在云母表面吸附后,改善了云母表面的疏水性,從而達到浮選目的.但長期以來,對于伯胺類浮選藥劑在云母表面的吸附現象沒有進行過系統研究,捕收劑對鋰云母的選擇性吸附行為及其吸附規律沒有系統理論研究,鋰云母精礦品位和浮選回收率都較低,精礦品位一般在3.8%左右,達不到優等品4.5%的目標,一級品4.0%的目標也難以達到,制約了企業的經濟效益,也對后續鋰云母深度開發產生不利影響.
云母是一種常見的層狀鋁硅酸鹽,因其(001)面解理后原子級的平整度好,且其表面天然是親水性的,因而常作為吸附基底用于研究界面現象. Nishimura等12使用AFM測定了烷基伯胺、仲胺、叔胺、季胺在云母表面的吸附,其半膠束濃度(HMC)隨取代基增加依次降低,而且其HMC和電位逆轉點(PZR)均隨堆積參數13增大而減小,而堆積參數實際受表面活性劑的碳鏈長度影響.2Fujii等3使用AFM測定除去過量吸附后的云母表面的單分子層CnTAB(即烷基鏈上碳原子數為n的三甲基溴化銨),結果證明吸附排布取決于表面晶格結構,與碳鏈的長度和數量無關.
對于硅酸鹽浮選體系研究,所采用的烷基胺的鏈長對浮選過程有很大影響.14-16無論是藥劑用量、起泡性能,還是吸附在礦石表面對其疏水性的改善效果等,都與所選用的烷基胺鏈長相關.為了研究吸附在云母表面的烷基伯胺的鏈長對其疏水性的影響規律,本文通過表面接觸角測量、原子力顯微鏡觀測、密度泛函理論和分子動力學模擬計算,比較了十二胺和十八胺兩種常用的表面活性劑在云母表面的吸附,以揭示不同烷基胺鏈長對浮選過程的影響,從而為藥劑的篩選及復配提供參考,并為提高鋰云母精礦品位和浮選回收率提供可能.
2 材料與方法
2.1 實驗材料
白云母片(Emsdiasum,V-1級),十二胺(CP)和鹽酸(AR)均購于國藥集團化學試劑有限公司,十八胺(Aldrich,90%),去離子水.十二胺和十八胺與鹽酸反應制備成相應的鹽酸鹽17后用于配制溶液.
將新鮮剝離的云母片浸入2×10-4mol·L-1的烷基伯胺鹽酸鹽中性溶液中,室溫下保持6 h時,取出后用去離子水洗滌30 s,用低速空氣吹除表面液滴,在干燥器中靜置24 h,然后分別用接觸角測量儀和原子力顯微鏡進行分析.接觸角使用上海中晨生產的JC2000D接觸角測量儀,按照量角法進行測量,在同一片云母片上取3個點進行測定.原子力顯微鏡采用美國Veeco公司NanoScopeIIIa multimode AFM的輕敲模式對云母片表面進行掃描得到表面圖像,用AFM掃描樣品表面均在室溫下(約20°C)進行,懸臂采用共振頻率為260-310 kHz、彈性系數為20-80 N·m-1的硅懸臂,AFM掃描的范圍為2.0 μm×2.0 μm,掃描頻率0.6-1 Hz.
2.2 計算模型與方法
采用Heinz等18,19加入層狀硅酸鹽內的原子勢能參數的PCFF_phyllosilicates力場(polymer consistent force field_phyllosilicates),它的半經驗模型加入了對表面能的考慮,計算的層狀硅酸鹽的晶體結構、表面能、振動頻率等均與實驗值18-20吻合.為驗證PCFF_phyllosilicates力場對云母計算結果的準確性,首先使用Accelrys Materials Studio 5.0軟件中的CASTEP模塊對單晶胞雙層白云母進行了DFT計算.云母是一種層狀硅酸鹽,每4個硅原子就有1個被鋁原子取代,云母在解理之后表面呈負電性(0.47 nm2·e-1),因此陽離子表面活性劑可以通過靜電力吸附到其表面,表面呈負電性鋁原子的取代從宏觀來看是無序的,而微觀上遵從Loewenstein規則.21選用的云母為2M1多型,晶胞參數的初始設定按照Kuwahara使用AFM測量的結果,22并割離出(001)面,建立一個5×6的云母超晶胞,尺寸為2.5959 nm× 2.7046 nm.鋁原子遵照Loewenstein規則部分取代云母表面的硅原子,使用Accelrys Materials Studio 5.0軟件中的Discover模塊進行結構優化.而單晶胞雙層云母的DFT計算選用GGA/PW91泛函,所有原子都按照模守恒贗勢(norm-conserving pseudopotential)計算,平面波最大截斷能設定為340 eV,最大殘余力為0.2 eV·nm-1,最大殘余應力為0.1 GPa.為了減少計算量,使用了虛擬晶體近似法(VCA)23,24來解決Al/Si的無序取代問題,鋁原子對硅原子的取代率為25%.
烷基伯胺分子和離子的結構使用Gaussian 03程序25DFT/B3LYP方法計算,采用6-31G(d)基組,根據計算得到的Mulliken電荷指派所有原子的電荷.水分子采用SPC/E模型26結構,在Accelrys Materials Studio 5.0軟件中使用rattle方法固定其鍵長和鍵角.
3 結果與討論
3.1 實驗結果
實驗測得在2×10-4mol·L-1的烷基伯胺鹽酸鹽中性溶液中,吸附了十二胺的云母表面接觸角為91°,吸附了十八胺的接觸角為94°,說明吸附了十八胺的云母疏水性略強于吸附了十二胺的云母.圖1是吸附了十二胺和十八胺的云母表面的AFM圖像.
圖1中,十二胺在云母表面形成了較為均勻的單層吸附,吸附厚度(最高值與最低值差值)為1.2-1.5 nm.考慮到十二胺分子的長度約為1.70 nm,推測十二胺是以一定夾角傾斜于云母表面所形成的單分子層吸附.十八胺在云母表面形成的吸附層厚度為3.9-4.4 nm,考慮到十八胺分子的長度為2.48 nm,推測十八胺以多分子層吸附的形式,從圖1中也可以看到十八胺吸附更為密集,并以球狀和短棒狀膠束的形式吸附在云母表面.
表面活性劑在固體表面的吸附形貌與其堆積參數有關,不同鏈長的烷基伯胺的堆積參數為v0/ al0,v0和l0分別是表面活性劑尾鏈的體積和長度,a是形成膠束時每個分子所占的疏水囊泡的表面積,可以通過公式(1)2求得:
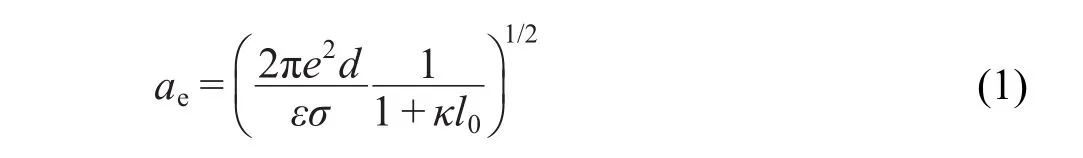
式(1)中,下標e代表其是臨界膠束濃度時數值,e是電子電量(4.8×10-10esu),d是雙電層模型的電容層厚度,ε是溶劑的相對介電常數(水:78.54),(2πe2d/ εσ)1/2=0.82 nm2,κ是德拜長度的相反數,由離子濃度決定:
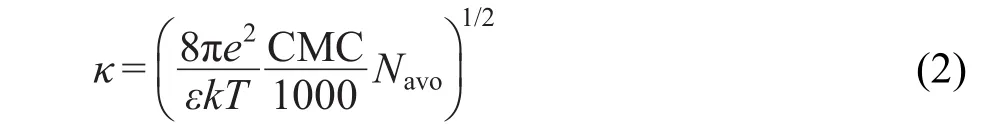
式(2)中k是波茲曼常數,T是溫度,CMC是表面活性劑臨界膠束濃度,十二胺和十八胺的CMC分別是1.2×10-2和1.5×10-4mol·L-1,Navo是阿伏伽德羅常數,從上式可以求得十二胺和十八胺的平衡堆積參數分別是0.1975和0.1673,所以二者不但在溶液中形成膠束的規律相近,而且在固體表面具有相近的吸附規律.在高于其CMC的溶液中,十二胺和十八胺都會形成球狀膠束,而在高于其HMC的時候,則會在固體表面吸附形成半膠束.根據Fuerstenau27的實驗結論,烷基伯胺的CMC大約是其HMC的100倍,實驗中配制的2×10-4mol·L-1濃度的溶液,略高于十二胺的HMC,遠高于十八胺的HMC.在溶液中烷基伯胺濃度相對于其HMC的倍數越大時,固體表面吸附的形貌變化按照四步吸附模型28由單分子層吸附向多層吸附變化,從而解釋了在該濃度下十八胺比十二胺更趨于形成多層吸附.
3.2 動力學計算
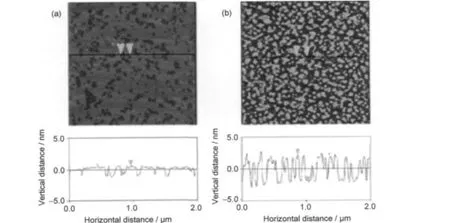
圖1 吸附十二胺(a)和十八胺(b)的云母表面的AFM圖像Fig.1 AFM images of the muscovite surface adsorbed with DDA(a)and ODA(b)
為了正確選擇MD計算所選用的力場,首先進行了力場的驗證.通過DFT計算和分子力學計算得到的2M1云母晶胞參數以及實驗值,22,29列于表1.其中解離能是指晶體的表面能γ,通過以下公式計算.

表1 實驗測定和不同方法計算的2M1云母晶胞參數和表面能(γ)Table 1 Cell parameters and cleave energy(γ)of 2M1 muscovite measured by experiments and calculated by different simulation methods

式中,Eslab是表面的總能量,Ebulk是體相能量,A是切面面積.
計算結果表明,PCFF_phyllosilicates力場對云母類礦石的計算較為準確,因此在MD計算中選用其作為力場.而對于DFT的能量計算而言可能單晶胞太小,所以得到的解離能誤差較大.
力場驗證計算過程中,優化后的十二胺陽離子和其分子相比,陽離子的極性基團(-CH2NH+3)的電荷由中性增大到0.814,使其可通過離子交換作用取代K+離子吸附到云母表面.而十二胺和十八胺的極性基團所帶電荷很相近,說明其伯胺基團和云母表面吸附位之間的庫侖(Coulomb)力受鏈長影響很小.實驗中所配制的十二胺鹽溶液pH范圍一般在5-8之間,此時十二胺主要以陽離子狀態存在.實際浮選生產操作中,不可能達到實驗室進行吸附實驗的條件要求,通常是將粗礦按30%-50%的固含率在常溫下進行調漿,很難形成穩定的單層吸附,這種情況下礦物表面的疏水性較實驗中要弱.

圖2 吸附十二胺的云母表面MD模擬的初始(a)和1 ns時(b)的結構截圖Fig.2 Snapshots of the initial state(a)and the state at 1 ns(b)of MD simulation for muscovite surface adsorbed with DDA
為了考察水溶液中十二胺陽離子向云母表面的吸附情況,構建了如圖2a所示的“三明治”形的體系(對氫原子進行了隱藏以更直觀),上下是對稱的5×6的2M1多型云母(001)面,中間是10 nm厚的含60個十二胺陽離子的水溶液.使用Discover模塊進行MD模擬,使用NVT系綜,采用Nose熱浴恒溫在298 K,步長為1 fs,范德華力截斷半徑為1 nm,庫侖力使用Ewald加和法計算,進行了1 ns的MD模擬后,體系仍未達到平衡,但原本隨機分布在溶液環境中的十二胺陽離子向兩端靠近云母表面的區域移動,結果如圖2b所示.
圖2b中,一部分十二胺陽離子極性基團已經吸附在云母表面,其碳鏈則相互吸引形成半膠束,排斥開原來在云母表面的水分子.溶液中的十二胺也團聚形成了較小的膠束.為了更直接地觀察十二胺向云母表面的吸附,以100 ps為一個時間段,分析其水密度平均分布的變化,如圖3所示.
圖3中,隨著時間推移,云母表面的水密度逐漸降低并接近于0,即原來隨機分布在溶液環境中的十二胺陽離子體現出向盒子兩端云母表面吸附的趨勢,并在500 ps后基本達到穩定狀態.因為系統是假定為中性的,沒有引入額外的水合氫離子,云母表面因為離子交換作用,部分鉀離子被表面活性劑陽離子端或水分子取代,即十二胺陽離子通過其頭基吸附在負電性的云母表面,其碳鏈占據了相當大比例的靠近云母表面區域的空間,使其體現出一定的疏水性.隨著時間的推移,表面形成更規則排布的單分子層、半膠束等結構,而這顯然是有利于浮選分離的結構形式.
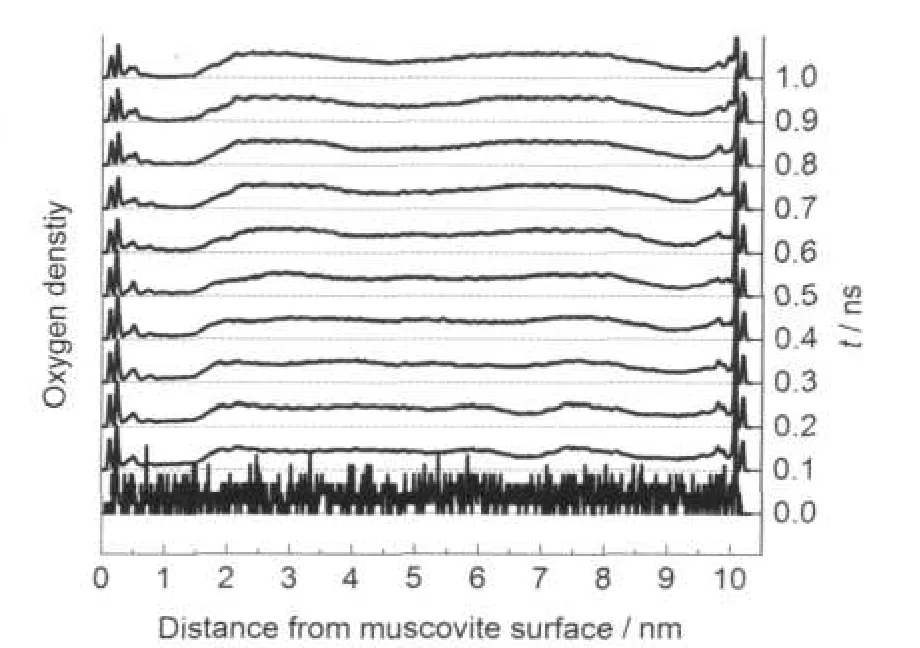
圖3 吸附十二胺的云母表面隨模擬時間變化的氧密度圖Fig.3 Oxygen density profile as a function of MD simulation time for muscovite surface adsorbed with DDA
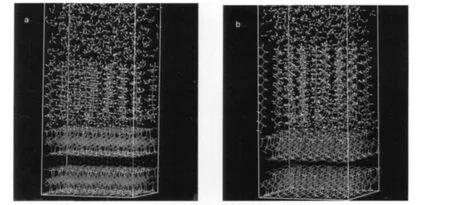
圖4 1 ns時云母表面截圖Fig.4 Snapshots of the muscovite surface at 1 ns (a)DDA-absorbed;(b)ODA-absorbed
由于烷基鏈的直徑(0.43-0.45 nm)比伯胺陽離子基團(0.37 nm)和伯胺基團(0.34 nm)都大,5,12而小于季胺陽離子基團(0.59 nm),12當烷基季胺陽離子在云母表面吸附時,除去表面過量吸附后,測得的占據面積約為0.23 nm2,3相當于K+離子交換面積的一半,即一個硅氧空穴的投影面積.因此,在烷基胺陽離子吸附并形成了規則的單分子層后,云母表面整體呈正電性,部分抗衡離子(如Cl-,Br-,CH3COO-等)會伴隨著吸附到靠近表面的區域以中和多余的正電荷.為了比較碳鏈長度對疏水性的影響,我們建立了單分子層的吸附模型,模型中包含30個烷基伯胺離子,取代了原來云母表面的K+離子作為其初始位置,豎直于對應的30個硅氧空穴上方,這相當于模擬Fujii等3使用AFM研究烷基胺在云母表面吸附形貌前對樣品的處理效果,即云母表面已經形成了規則排布的單分子層并去除了多余的烷基胺,此時表面呈最大的疏水性.將真空環境內的體系進行能量最小化計算,并進行1 ns的MD模擬后,兩種烷基伯胺均以傾斜一定角度的單分子層形式吸附在云母表面.在烷基伯胺單分子層上方的真空層加入水相,密度為1 g·cm-3.MD模擬的設置與前文相同,又進行了1 ns的MD模擬之后,體系能量、溫度已經達到平衡,再進行平衡時間為100 ps的MD模擬,數據采集的頻率是100 fs,取其平均值用于分析.兩個模型在1 ns時的云母表面截圖如圖4所示.
從圖4中可以看出,少量的水分子進入原本有序排列的烷基伯胺單分子層內,使其排布受到一定影響,同時混合使體相水密度也發生了一定變化,所有兩個體系在平衡時水密度略有不同,十二胺體相水平均密度為0.894,十八胺體系則為0.925.因此在后面的討論中也以水密度與體相平均密度之比作為依據來討論.
疏水性強弱可以通過靠近表面區域的水密度進行比較.30一個表面的疏水性越強,宏觀上接觸角越大,微觀上氧原子密度的第一個峰數值越小.因為初始狀態下的烷基伯胺單分子層并不是其最穩定的吸附形式,所以有一定量的水分子會進入單分子層的碳鏈間距之間,但整體上未破壞單分子層的結構,仍然體現出與Trudeau等30研究固體表面上疏水性相同的趨勢.計算的十二胺和十八胺在云母表面吸附后沿云母(001)面向外的氧密度見圖5.
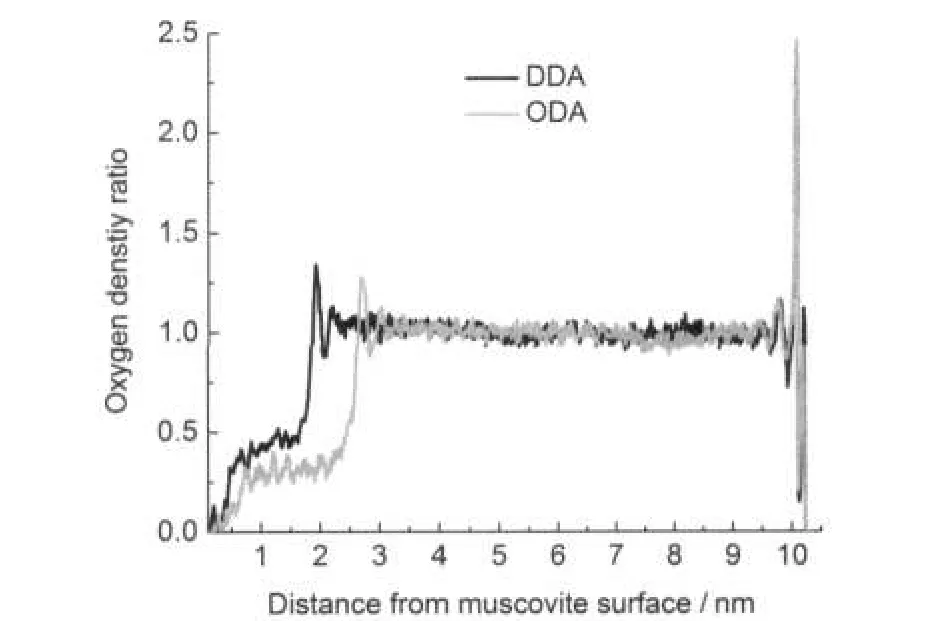
圖5 沿云母表面向外的氧密度圖Fig.5 Oxygen density profile as a function of the distance from the muscovite surfaceoxygen density ratio:ratio of oxygen density at a place to average oxygen density in the bulk
圖5中,氧密度在十二胺體系中出現的第一個峰距離云母表面的距離為1.91 nm,十八胺的第一個峰距離為2.68 nm,而相應的十二胺和十八胺單分子層厚度則約為1.682和2.463 nm,這其中還包括了摻入的水分子所占據的體積.十二胺體系的氧密度峰值為1.200,氧密度峰值和體相之比為1.341,高于十八胺的1.185和1.284,說明十二胺單層膜的疏水性比十八胺弱,與接觸角測量的結果一致.而圖5右側出現的水在云母表面吸附出現的高峰,體現了云母本身的親水性.另外,考慮到實驗中十八胺的吸附形式并非是疏水性最強的單分子層結構,所以在相同吸附狀態下,吸附了十二胺和十八胺的云母疏水性差異應比本實驗中接觸角測量的結果差別更大.
為了進一步考察界面上的氫鍵情況,定義分子間氫鍵成鍵距離小于0.25 nm.在此基礎上,計算了平均氫鍵數量,寬度為0.01 nm,結果見圖6.
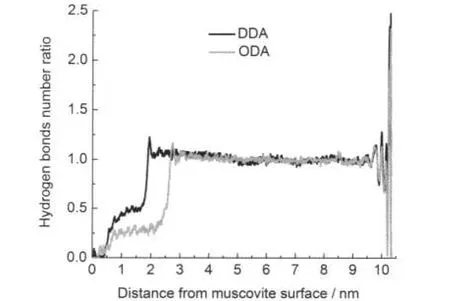
圖6 沿云母表面向外的氫鍵數量分布圖Fig.6 Hydrogen bonds number profile as a function of the distance from the muscovite surfacehydrogen bonds number ratio:ratio of hydrogen bonds number at a place to average hydrogen bonds number in the bulk
4 結論
本文使用PCFF_phyllosilicates力場對烷基伯胺在云母表面吸附進行了MD模擬,發現烷基伯胺可以通過范德華力和氫鍵作用吸附在云母表面.由于碳氫鏈尾對水分子的排斥,使云母由親水性轉化為疏水性.通過比較氧密度和氫鍵數量分布,研究了十二胺和十八胺單分子層吸附狀態下的表面疏水性,碳鏈越長,其對云母表面疏水性改善的能力越強,計算結果與實驗測得的接觸角吻合.
因為十八胺的CMC和HMC要遠低于十二胺的,所以具有更好的起泡性能,也更易在云母表面形成多層吸附.在2×10-4mol·L-1濃度下,十八胺以球狀和短棒狀膠束的形式吸附在云母表面,其接觸角仍比十二胺單層吸附的云母要大.在浮選生產等實際應用中,長鏈表面活性劑能在更低濃度下更好地改善固體表面的疏水性,但同時隨著碳鏈的增長,其水溶性也會有所下降.因此,在浮選藥劑復配中,應在考慮水溶性的基礎上,盡量使用同系物中的長鏈表面活性劑以提高浮選效果和減少浮選藥劑的用量.
(1)Patrick,H.N.;Warr,G.G.;Manne,S.;Aksay,I.A.Langmuir 1999,15,1685.
(2)Nagarajan,R.Langmuir 2001,18,31.
(3)Fujii,M.;Li,B.Y.;Fukada,K.;Kato,T.;Seimiya,T.Langmuir 2001,17,1138.
(4)Patil,K.G.;Santhanam,V.;Biswas,S.K.;Ayappa,K.G. J.Phys.Chem.C 2010,114,3549.
(5)Vidyadhar,A.;Rao,K.H.;Chernyshova,I.V.Colloid Surf. A-Physicochem.Eng.Asp.2003,214,127.
(6)Wang,X.M.;Liu,J.;Du,H.;Miller,J.D.Langmuir 2010,26, 3407.
(7) Du,H.;Miller,J.D.Int.J.Miner.Process.2007,84,172.
(8)Zehl,T.;Wahab,A.;Schiller,P.;Mogel,H.J.Langmuir 2009, 25,2090.
(9)Zhang,R.;Liu,C.;Somasundaran,P.J.Colloid Interface Sci. 2007,310,377.
(10)Liu,X.Y.;Li,C.;Tian,W.Y.;Chen,T.;Wang,L.H.;Zheng,Z.; Zhu,J.B.;Sun,M.;Liu,C.L.Acta Phys.-Chim.Sin.2011,27, 59.[劉曉宇,黎 春,田文宇,陳 濤,王路化,鄭 仲,朱建波,孫 茂,劉春立.物理化學學報,2011,27,59.]
(11) Song,Q.S.;Guo,X.L.;Yuan,S.L.;Liu,C.B.Acta Phys.-Chim.Sin.2009,25,1053. [宋其圣,郭新利,苑世領,劉成卜.物理化學學報,2009,25,1053.]
(12)Nishimura,S.;Scales,P.J.;Biggs,S.;Healy,T.W.Langmuir 2000,16,690.
(13) Israelachvili,J.N.;Mitchell,D.J.;Ninham,B.W.J.Chem. Soc.Faraday Trans.2 1976,72,1525.
(14)Pugh,R.J.;Rutland,M.W.;Manev,E.;Claesson,P.M.Int.J. Miner.Process.1996,46,245.
(15) Zhang,R.;Somasundaran,P.Adv.Colloid Interface Sci.2006, 123,213.
(16)Jiang,H.;Hu,Y.H.;Tan,W.Q.;Wang,Y.H.;Wang,D.Z.Chin. J.Nonferrous Met.2001,11,688. [蔣 昊,胡岳華,覃文慶,王毓華,王淀佐.中國有色金屬學報,2001,11,688.]
(17)Kong,Y.X.;Di,Y.Y.;Zhang,Y.Q.;Yang,W.W.;Tan,Z.C. Thermochim.Acta 2009,495,33.
(18)Heinz,H.;Koerner,H.;Anderson,K.L.;Vaia,R.A.;Farmer,B. L.Chem.Mat.2005,17,5658.
(19)Heinz,H.;Vaia,R.A.;Farmer,B.L.;Naik,R.R.J.Phys.Chem. C 2008,112,17281.
(20)Heinz,H.;Vaia,R.A.;Farmer,B.L.J.Chem.Phys.2006,124, 224713.
(21)Loewenstein,W.Am.Miner.1954,39,92.
(22)Kuwahara,Y.Phys.Chem.Miner.1999,26,198.
(23) Bellaiche,L.;Vanderbilt,D.Phys.Rev.B 2000,61,7877.
(24)Winkler,B.;Pickard,C.;Milman,V.Chem.Phys.Lett.2002, 362,266.
(25) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, Revision D.01;Gaussian Inc.:Pittsburgh,PA,2003.
(26) Berendsen,H.J.C.;Grigera,J.R.;Straatsma,T.P.J.Phys. Chem.1987,91,6269.
(27) Fuerstenau,D.J.Phys.Chem.1956,60,981.
(28) Fan,A.;Somasundaran,P.;Turro,N.J.Langmuir 1997,13,506.
(29)Heinz,H.;Suter,U.W.J.Phys.Chem.B 2004,108,18341.
(30)Trudeau,T.G.;Jena,K.C.;Hore,D.K.J.Phys.Chem.C 2009, 113,20002.
July 18,2011;Revised:October 26,2011;Published on Web:October 31,2011.
Effect of Primary Alkylamine Adsorption on Muscovite Hydrophobicity
LIU Zhen LIU Gou-Sheng*YU Jian-Guo
(State Key Laboratory of Chemical Engineering,East China University of Science and Technology,Shanghai 200237,P.R.China)
The adsorption of surfactants on mineral surface has a great influence on the solid hydrophobicity and flotation behavior.The relationship between the hydrocarbon tail length of the primary alkylamines and muscovite hydrophobicity was investigated by contact angle measurement,atomic force microscopy(AFM),density functional theory(DFT),and molecular dynamics(MD)simulation.By comparing the oxygen density and the hydrogen bonds number profile,we observed that the formed hydrogen bonds for each water molecule on the interface between hydrocarbon tails and the water phase were fewer than that in the bulk.Additionally,the muscovite that absorbed alkylamines transformed from a hydrophilic surface to hydrophobic one.We also found that the octadecylamine(ODA)-absorbed muscovite surface was more hydrophobic than the dodecylamine(DDA)-absorbed surface while they were both in a monolayer state.Furthermore,because octadecylamine has a much lower hemi-micelle concentration (HMC)than dodecylamine,it forms multilayer more easily,meaning that the primary alkylamine with longer hydrocarbon tail is a better choice for the hydrophobicity enhancement of muscovite surface.The experimental results are in good agreement with theoretical calculations.
Muscovite;Adsorption;Hydrophobicity;Atomic force microscopy;Molecular dynamics simulation
10.3866/PKU.WHXB201228201
*Corresponding author.Email:gsliu@ecust.edu.cn;Tel/Fax:+86-21-64250981.
The project was supported by the National Natural Science Foundation of China(51164009).
國家自然科學基金(51164009)資助項目
O647
