聚乙烯醇縮丁醛準固態電解質薄膜的制備和性能表征
2012-12-21 06:33:42黃其煜王曉晨齊芳藝鄭一胄
物理化學學報 2012年5期
周 偉 黃其煜,* 王曉晨 齊芳藝 焦 方 鄭一胄
(1上海交通大學微電子學院,上海200240;2上海交通大學材料科學與工程學院,上海200240)
聚乙烯醇縮丁醛準固態電解質薄膜的制備和性能表征
周 偉1黃其煜1,*王曉晨2齊芳藝1焦 方1鄭一胄1
(1上海交通大學微電子學院,上海200240;2上海交通大學材料科學與工程學院,上海200240)
研究了聚乙烯醇縮丁醛準固態電解質薄膜的制備及相關性能.通過向聚乙烯醇縮丁醛中加入適量造孔劑和輔助劑制備電解質薄膜,研究了薄膜制備過程中的相關影響因素和不同孔隙率的電解質薄膜對電池光電轉換效率的影響.實驗表明,通過向0.200 g聚乙烯醇縮丁醛中加入6.000 g碳酸鈣、0.310 g氯化鈣和0.150 g葡萄糖所制備的電解質薄膜性能最優,用其制備的染料敏化太陽能電池光電轉換效率η=4.720%(開路電壓Voc=0.7194 V,短路電流密度Jsc=10.014 mA·cm-2,填充因子FF=0.6559),達到相同條件下液態電解質電池的88%以上.薄膜電解質制備簡單,封裝方便,所用原料無毒無害,具有一定的發展潛力.
染料敏化太陽能電池;準固態電解質;電解質薄膜;聚乙烯醇縮丁醛;多孔膜
1 引言
染料敏化太陽能電池(DSSCs)是近年來發展興起的一種新型太陽能電池,由于其具有原料價廉易得、制造工藝簡單環境友好、在非直射和弱光環境中仍能高效工作等優點,從1991年首次報道以來便備受矚目,成為光伏研究中的熱點.1它被認為是傳統硅基太陽能電池最具潛力的替代方案.經過近二十年的研究改進,實驗室目前研究的染料敏化太陽能電池光電轉換效率已超過11%,2與傳統多晶硅太陽能電池之間光電轉換效率差距正在不斷減小.隨著商業化前景日益顯現,很多公司也投入巨資積極推進應用研究.3,4
染料敏化太陽能電池主要由光陽極、電解質和對電極組成.電解質的物理性質很大程度上決定著電池的封裝形式,因為液態電解質電池難以封裝容易出現漏液,這同時也限制了電池成品的形狀.而現階段固態電解質染料敏化太陽能電池光電轉換性能低下,5與液態電解質光電轉換效率還有很大差距,于是準固態電解質成為了現階段實用化研究的一個熱點.通常,準固態電解質是通過向液體電解質中加入有機小分子、6,7高聚物8,9或納米顆粒10,11使液體電解質凝膠化制得,然而這種制備方法所獲得的準固態電解質為布丁狀,在一定程度上仍然存在封裝困難的問題.
為解決這一問題,本文提出使用聚乙烯醇縮丁醛制備薄膜態準固態電解質.聚乙烯醇縮丁醛無毒無害,具有很高的光透射率和化學穩定性,12,13耐寒、耐紫外線和耐老化性能優良,同時絕緣性好,14,15對玻璃等有良好的粘結力,被廣泛應用于制造夾層玻璃(如安全玻璃)的中間夾層和涂料,16,17近年來其優良特性被不斷發掘利用.18-21此外,聚乙烯醇縮丁醛薄膜制備過程無污染,無需加熱、加壓等條件,可以有效降低能源消耗,實現綠色生產.本文對相關制備過程及影響因素進行了研究.
2 制備原理
聚乙烯醇縮丁醛不溶于水,易溶于乙醇,12形成黏度較大的膠體.向膠體中加入適量造孔劑攪拌均勻,刮涂在玻璃表面,待乙醇揮發在玻璃表面便形成一層薄膜.選擇合適的方式除去薄膜中的造孔劑形成透明聚乙烯醇縮丁醛多孔薄膜,將薄膜浸入液態電解質中,通過薄膜中的微孔腔吸納電解質溶液微液滴,構成離子遷移的通道.22,23同時,聚乙烯醇縮丁醛絕緣性能優良,可有效防止封裝時兩電極間接觸短路.
3 實驗方案
3.1 光陽極的制備
將TiO2漿料(中科聚鑫太陽能科技有限責任公司)通過絲網印刷在FTO導電玻璃上制備一層10 μm左右的薄膜,24TiO2顆粒直徑約為25 nm,將薄膜放入電爐中加熱到500°C保溫1 h并隨爐冷卻至室溫,再將其放入0.05 mol·L-1的TiCl4溶液(國藥集團化學試劑有限公司,分析純.本文所用化學試劑除特別標明外均購置于此公司,且均為分析純),水浴加熱30 min,25水浴溫度恒定為65°C,取出用去離子水洗凈晾干再放入電爐加熱到500°C保溫1 h,隨爐冷卻至80°C立即浸入0.3×10-3mol·L-1N719染料(大連七色光太陽能科技開發公司)溶液中,26浸泡18 h后取出用無水乙醇洗凈晾干備用.制得的二氧化鈦多孔薄膜表面形貌如圖1所示(未浸泡染料前).
3.2 準固態電解質薄膜的制備
取0.200 g聚乙烯醇縮丁醛(國藥集團化學試劑有限公司,航空級(滬試)),加入適量CaCO3粉體(顆粒直徑1-5 μm)作為造孔劑,并加入一定量無水CaCl2和葡萄糖作為輔助劑,攪拌均勻后加入適量無水乙醇攪拌成合適黏度的糊狀體,超聲30 s(上海三友超音設備有限公司,D68H)去除膠體中的氣泡.用刮板法將所得糊狀物刮涂在載玻片表面,待無水乙醇揮發后便在載玻片表面形成一層薄膜.由于聚乙烯醇縮丁醛機械強度較高,薄膜可以很容易地從載玻片表面剝離.將取下的薄膜浸入稀鹽酸溶液中.若干小時后,薄膜中的CaCO3鹽酸反應完全,溶液中留下完全透明的聚乙烯醇縮丁醛多孔薄膜.向溶液中加入一定量NaOH溶液中和至中性,再將薄膜取出用大量去離子水沖洗、浸泡晾干.將晾干的聚乙烯醇縮丁醛多孔膜浸入液態電解質(中科聚鑫太陽能科技有限公司,CJX-ES-10)中,待吸納完全后取出薄膜,用濾紙吸去薄膜表面的電解質液體.
3.3 電池的封裝

圖1 納米晶二氧化鈦薄膜表面形貌Fig.1 Surface morphology of nanocrystalline titanium dioxide thin film
從電解質薄膜上裁取光陽極上二氧化鈦納米薄膜大小的一塊平鋪在二氧化鈦薄膜表面,將濾紙(厚度約為160 μm)剪成“回”字型蓋在光陽極上,使TiO2薄膜處在“回”字型的中部,蓋上對電極(大連七色光太陽能科技開發有限公司)并壓緊,吸去多余的電解質溶液,在中間縫隙處涂上光敏樹脂并在紫外燈下曝光固化,至此整個電池封裝完畢.
4 結果及討論
4.1 聚乙烯醇縮丁醛薄膜透射率
本文采用雙光束紫外-可見分光光度計(北京普析通用儀器有限責任公司,TU-1901)測量聚乙烯醇縮丁醛薄膜的透射率(薄膜厚度約400 μm),結果如圖2所示.在所測量的400-900 nm波長范圍內,薄膜的透射率接近90%.
4.2 輔助劑的含量對薄膜制備的影響
取0.200 g聚乙烯醇縮丁醛,加入5.000 g CaCO3,再分別加入一定量的CaCl2和葡萄糖,按上述方法在載玻片刮涂薄膜,晾干后浸入稀鹽酸溶液,記錄薄膜完全透明所需要的時間,結果如表1所示.
表1中,“-”表示加入該含量的添加劑所調制的糊狀物不能很好成膜而終止實驗:加入0.420 g CaCl2使得糊狀物中形成大量的顆粒物;加入0.250 g葡萄糖降低糊狀物的黏度使刮涂的薄膜不連續.由表1可知,加入0.310 g CaCl2時,去除薄膜中的CaCO3所需時間最短,葡萄糖的含量對去除造孔劑的時間影響相對較弱,當其含量達到0.195 g時,所制得薄膜的強度明顯下降.根據以上結果,本文認為最優的輔助劑含量為0.310 g CaCl2和0.150 g葡萄糖.
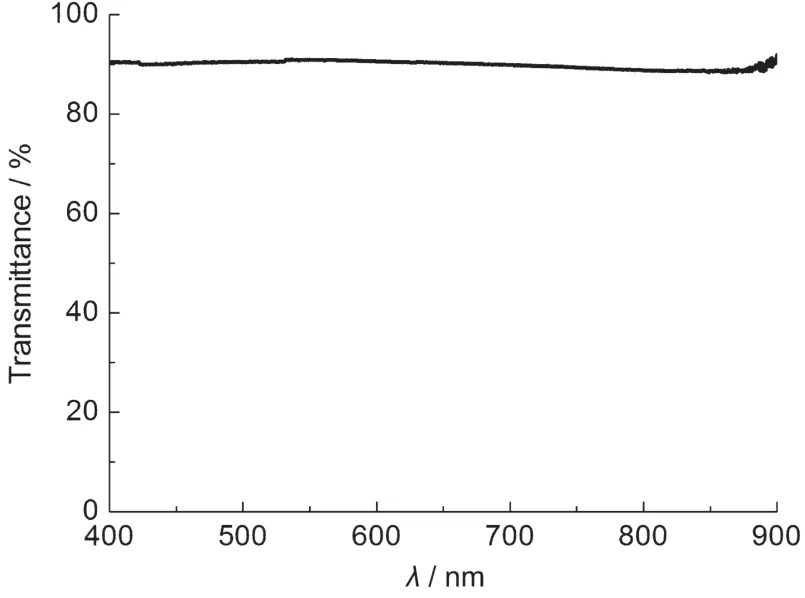
圖2 聚乙烯醇縮丁醛薄膜透射率Fig.2 Transmittance of polyvinyl butyral film
CaCl2和葡萄糖的加入不僅能加速造孔劑的除去,同時可以降低刮涂的薄膜與載玻片之間的粘結,可以更容易地將薄膜從載玻片上完整剝離,這可能與這兩種物質會在空氣中吸收水分形成液滴潤濕薄膜有關,如圖3所示,左側薄膜中未加入輔助劑,右側樣品中加入輔助劑,在空氣中靜置,右側樣品產生小液滴.
4.3 造孔劑含量對吸納液態電解質的影響
取0.200 g聚乙烯醇縮丁醛,分別加入4.000、5.000、6.000和7.000 g CaCO3,再加入0.310 g CaCl2和0.150 g葡萄糖,刮制薄膜并除去造孔劑.用分析天平(METTLER TOLEDO,XS105)稱量最后所得透明薄膜的干重,將透明薄膜浸入液態電解質中,待薄膜完全吸納電解質液體后取出,吸去表面多余電解質溶液,測量浸泡液態電解質后的濕重;再將23.523 g重物壓在薄膜上,吸去析出的電解質溶液,測量此刻薄膜濕重.分別計算濕重與干重的比值,結果如圖4所示.

圖3 輔助劑對薄膜形貌的影響Fig.3 Effect of auxiliaries on the morphologies of films(a)without auxiliaries,(b)with auxiliaries; noting the liquid drops forming on glass
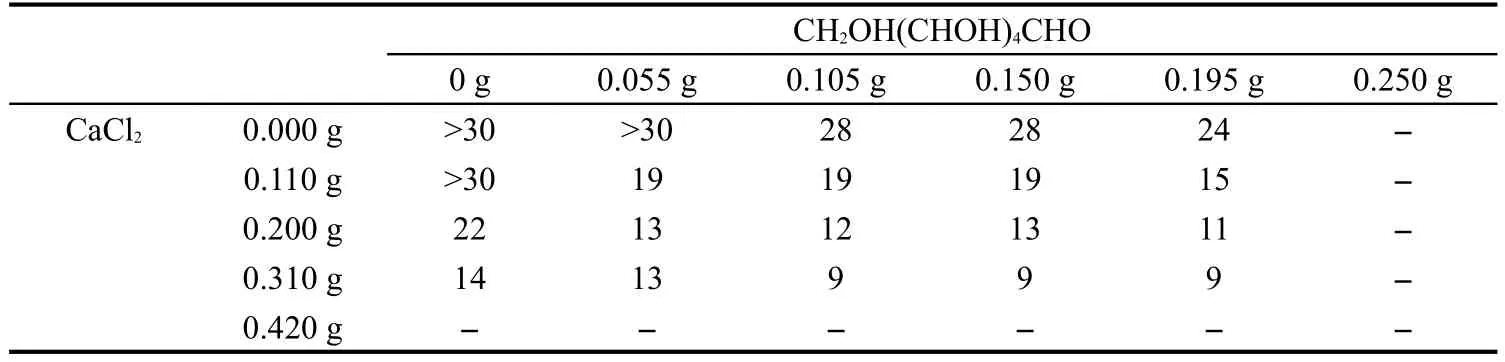
表1 不同含量輔助劑對造孔劑除去時間(單位為h)的影響Table 1 Time(unit in h)to remove pore forming agent of films with different levels of auxiliaries
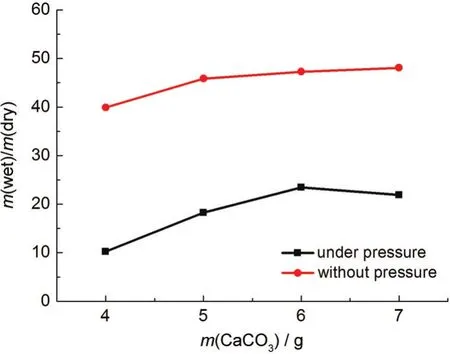
圖4 造孔劑含量對薄膜吸納電解質溶液的影響Fig.4 Impact of different contents of pore forming agent on the absorption of electrolyte by films
由圖4可知,在未對薄膜施加外加壓力之前,聚乙烯醇縮丁醛薄膜吸附液態電解質的質量是薄膜本身質量的40-50倍,展現了聚乙烯醇縮丁醛多孔薄膜良好的吸納電解質的能力.將23.523 g重物壓在薄膜上,液態電解質的質量仍是其自身質量的10-23倍.同時可以看出,隨著造孔劑CaCO3含量的增加,多孔薄膜吸納電解質溶液的質量也在不斷增加,但增加的幅度趨緩;在外加作用力后,加6.000 g CaCO3的多孔膜具有最強的電解質溶液的保持能力,造孔劑含量太多不利于電解質溶液的保持.
4.4 溫度對造孔劑除去的影響
取0.200 g聚乙烯醇縮丁醛,加入6.000 g CaCO3、0.310 g CaCl2和0.150 g葡萄糖攪拌均勻,并適量加入無水乙醇刮涂成薄膜.將薄膜放入不同濃度稀鹽酸溶液中浸泡,記錄完全去除CaCO3所需的時間,溫度對除去造孔劑所需時間的影響如圖5所示.
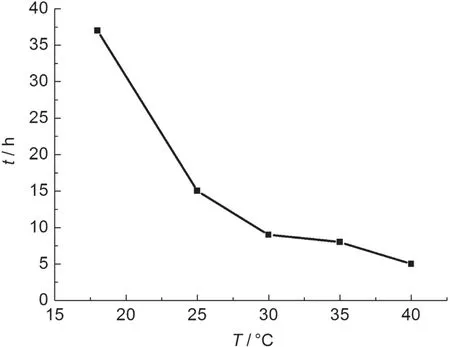
圖5 溫度對除去造孔劑所需時間的影響Fig.5 Time needed to remove pore forming agent under different temperatures
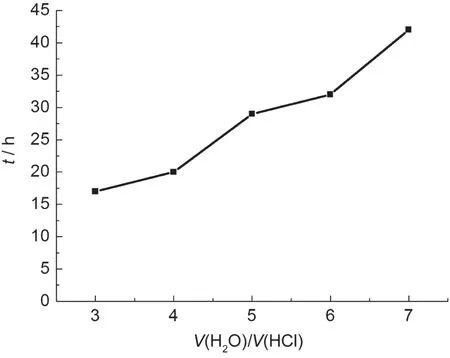
圖6 不同鹽酸濃度下去除造孔劑所需時間Fig.6 Time needed for the removal of pore forming agent with different concentrations of hydrochloric acid
由圖5可知,溫度對去除造孔劑所需時間影響較大,溫度越高造孔劑除去速率越快.實際實驗中發現,隨著溫度的升高,去除造孔劑后所得的多孔聚乙烯醇縮丁醛薄膜的強度逐漸降低,這可能與碳酸鈣與鹽酸反應速率過快生成的二氧化碳氣體在小孔內積聚膨脹破壞孔洞之間的粘連有關.因此,選擇在30°C恒溫下完成薄膜制備.
4.5 鹽酸濃度對造孔劑除去所需時間的影響
取0.200 g聚乙烯醇縮丁醛,加入6.000 g CaCO3、0.310 g CaCl2和0.150 g葡萄糖刮制成薄膜.取4 mL濃鹽酸(國藥集團化學試劑有限公司,分析純,質量分數37%)并按一定體積比加入去離子水,配制成不同濃度的稀鹽酸溶液.將制得的薄膜浸入鹽酸溶液,記錄造孔劑被完全去除所需的時間(22°C),不同鹽酸濃度下去除造孔劑所需時間如圖6所示.
隨著鹽酸濃度的降低,去除造孔劑所需的時間快速增長.當去離子水與濃鹽酸的體積比小于3時,配制出的溶液揮發性較高,通常采用體積比為3的溶液去除造孔劑,盡量縮短實驗所需時間.
4.6 造孔劑含量對電池光電轉換效率的影響
取0.200 g聚乙烯醇縮丁醛,分別加入4.000、5.000、6.000和7.000 g CaCO3,再加入0.310 g CaCl2、0.150 g葡萄糖和適量無水乙醇按上述方法制備準固態電解質,并封裝成染料敏化太陽能電池進行測試.作為比較同時制備了純液態電解質染料敏化太陽能電池.測試結果如表2所示.
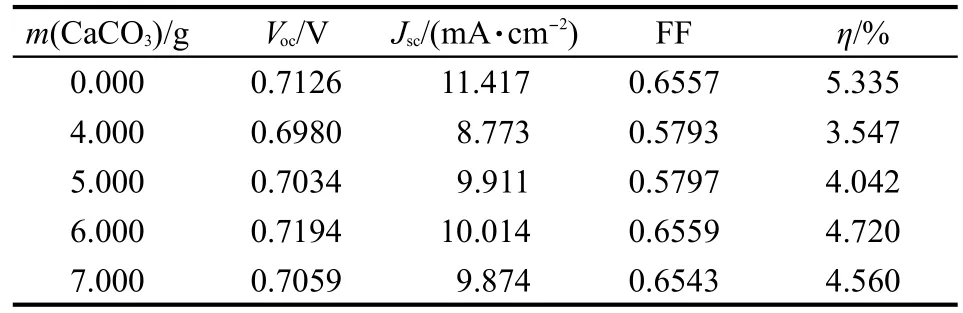
表2 含有不同造孔劑質量的準固態電解質薄膜電池測試結果Table 2 Testing results of quasi-solid state solar cells with different contents of pore forming agent
由表2可知,在造孔劑含量較少時,離子傳輸通道較少,光電轉換效率較低,當造孔劑含量達到6.000 g時,光電轉換效率達到最大,達到相同條件下純液態電解質染料敏化太陽能電池光電轉換效率(η=5.335%)的88%以上,進一步增大造孔劑的含量使得薄膜在外界作用下保持液態電解質溶液的能力減弱,在封裝擠壓時電解質流失,光電裝換效率略有降低.
5 結論
采用聚乙烯醇縮丁醛制備準固態電解質薄膜,研究結果表明:在0.200 g聚乙烯醇縮丁醛中,(1)加入0.310 g CaCl2和0.150 g葡萄糖并在30°C時除去碳酸鈣造孔劑所需要的時間最短(約為9 h),性能最優;(2)隨著CaCO3造孔劑含量的增加薄膜吸納電解質溶液的能力逐漸增強,當CaCO3造孔劑含量為6.000 g時,所制備的電解質薄膜在外加壓力下有著最好的電解液保持性能;(3)在造孔劑含量為6.000 g時,封裝所得的準固態電解質染料敏化太陽能電池的光電轉換效率最高為4.720%,達到相同條件下純液態染料敏化太陽能電池的88%以上.綜上,這種準固態電解質薄膜制備方法簡單,所制備出的薄膜形態電解質易于封裝,同時對電池光電轉換效率影響較小,具有一定的發展潛力.
(1)O'Regan,B.;Gr?tzel,M.Nature 1991,353(6346),737.
(2)Gr?tzel,M.Comptes Rendus Chimie 2006,9(5-6),578.
(3)Kong,F.T.;Dai,S.Y.;Wang,K.J.Advances in OptoElectronics 2007,2007,75384.
(4)Yu,Z.X.;Li,D.M.;Qin,D.;Sun,H.C.;Zhang,Y.D.;Luo,Y. H.;Meng,Q.B.Materials China 2009,7(8),8.[于哲勛,李冬梅,秦 達,孫惠成,張一多,羅艷紅,孟慶波.中國材料進展, 2009,7(8),8.]
(5) Jiang,X.;Karlsson,K.M.;Gabrielsson,E.;Johansson,E.M.J.; Quintana,M.;Karlsson,M.;Sun,L.;Boschloo,G.;Hagfeldt,A. Advanced Functional Material 2011,21(15),2944.
(6)Huo,Z.P.;Zhang,C.N.;Fang,X.Q.;Cai,M.L.;Dai,S.Y.; Wang,K.J.Journal of Power Sources 2010,195(13),4384.
(7)Huo,Z.P.;Dai,S.Y.;Zhang,C.N.;Liu,W.Q.;Fang,X.Q.; Cai,M.L.;Guo,L.;Wang,K.J.;Jiang,N.Q.;Zheng,Y.Z. Chem.J.Chin.Univ.2009,30(6),1214.[霍志鵬,戴松元,張昌能,劉偉慶,方霞琴,蔡墨朗,郭 磊,王孔嘉,姜年權,鄭亦莊.高等學校化學學報,2009,30(6),1214.]
(8)Kim,J.Y.;Kim,T.H.;Kim,D.Y.;Park,N.G.;Ahn,K.D. Journal of Power Sources 2008,175(1),692.
(9)Yu,Z.X.;Da,Q.;Zhang,Y.D.;Sun,H.C.;Luo,Y.H.;Meng, Q.B.;Li,D.M.Energy Environ.Sci.2011,4(4),1754.
(10)Wang,P.;Zakeeruddin,S.M.;Comte,P.;Exnar,I.;Gr?tzel,M. Journal of the American Chemical Society 2003,125(5),1166.
(11) Sharma,G.;Suresh,P.;Roy,M.;Mikroyannidis,J.A.Journal of Power Sources 2010,195(9),3011.
(12) Tong,Y.;Liu,Y.;Lu,S.;Dong,L.;Chen,S.;Xiao,Z.Journal of Sol-Gel Science and Technology 2004,30(3),157.
(13) Yuan,H.L.;Ma,P.L.;Wang,J.;Li,J.;Guo,X.Plastics Science and Technology 2004,32(2),4.[苑會林,馬沛嵐,王 婧,李 軍,郭 嫻.工程塑料應用,2004,32(2),4.]
(14)Dong,L.J.;Xiong,C.X.;Quan,H.Y.;Zhao,G.H.Scripta Materialia 2006,55(9),835.
(15) Jain,K.;Kumar,N.;Mehendru,P.Journal of the Electrochemical Society 1979,126(11),1958.
(16)Dhaliwal,A.;Hay,J.Thermochimica Acta 2002,391(1-2), 245.
(17) Salam,L.A.;Matthews,R.D.;Robertson,H.Journal of the European Ceramic Society 2000,20(9),1375.
(18)Zhu,H.B.;Liu,Z.C.;Kong,D.J.;Wang,Y.D.;Xie,Z.K.The Journal of Physical Chemistry C 2008,112(44),17257.
(19)Yuan,R.;Liu,Y.;Li,Q.F.;Chai,Y.Q.;Mo,C.L.;Zhong,X.; Tang,P.P.;Dai,J.Y.Analytical and Bioanalytical Chemistry 2005,381(3),762.
(20) Zhang,Y.B.;Ding,Y.P.;Gao,J.Q.;Yang,J.F.Journal of the European Ceramic Society 2009,29(6),1101.
(21) Roszol,L.;Lawson,T.;Koncz,V.;Noszticzius,Z.;Wittmann, M.;Sarkadi,T.;Koppa,P.The Journal of Physical Chemistry B 2010,114(43),13718.
(22) Li,H.Q.;Zhang,Y.Z.;He,P.;Li,H.;Li,R.Journal of Tianjin Polytechnic University 2005,24(5),37.[李慧琴,張玉忠,賀 鵬,李 泓,李 然.天津工業大學學報,2005,24(5),37.]
(23)Sheng,F.;Lu,X.F.;Shi,L.Q.;Bian,X.K.Membrane Science and Technology 2006,26(6),6.[沈 飛,陸曉峰,施柳青,卞曉鍇.膜科學與技術,2006,26(6),6.]
(24) Fredin,K.;Johansson,E.M.J.;Blom,T.;Hedlund,M.; Plogmaker,S.;Siegbahn,H.;Leifer,K.;Rensmo,H.Synthetic Metals 2009,159(1-2),166.
(25)Chen,D.P.;Zhang,X.D.;Wei,C.C.;Liu,C,C.;Zhao,Y.Acta Phys.-Chim.Sin 2011,27(2),425.[陳東坡,張曉丹,魏長春,劉彩池,趙 穎.物理化學學報,2011,27(2),425.]
(26)Paulose,M.;Shankar,K.;Varghese,O.K.;Mor,G.K.;Hardin, B.;Grimes,C.A.Nanotechnology 2006,17(49),1446.
December 5,2011;Revised:February 21,2012;Published on Web:March 5,2012.
Fabrication and Characterization of Quasi-Solid-State Electrolyte Films Based on Polyvinyl Butyral
ZHOU Wei1HUANG Qi-Yu1,*WANG Xiao-Chen2QI Fang-Yi1JIAO Fang1ZHENG Yi-Zhou1
(1School of Microelectronics,Shanghai Jiao Tong University,Shanghai 200240,P.R.China;2School of Materials Science and Engineering,Shanghai Jiao Tong University,Shanghai 200240,P.R.China)
The preparation and properties of quasi-solid-state electrolyte films based on polyvinyl butyral were studied.The films were prepared by adding a pore forming agent and auxiliary agents to polyvinyl butyral.Factors affecting the preparation of the films were investigated.The results showed that the films prepared by adding 6.000 g calcium carbonate,0.310 g calcium chloride,and 0.150 g glucose to 0.200 g polyvinyl butyral performed best.The impact of different pore ratios on device efficiency was investigated as well.The efficiency of dye-sensitized solar cells(DSSCs)using the quasi-solid-state electrolyte film reached 4.720%(open-circuit voltage Voc=0.7194 V,short-circuit current density Jsc=10.014 mA·cm-2,fill factor FF=0.6559),which was 88%of that for the corresponding liquid electrolyte solar cells.The film is easy to use for packaging and its preparation method is simple and non-toxic.
Dye-sensitized solar cell; Quasi-solid state electrolyte;Electrolyte film; Polyvinyl butyral;Porous membrane
10.3866/PKU.WHXB201203051
?Corresponding author.Email:qiyu@sjtu.edu.cn;Tel:+86-21-34204546.
The project was supported by the Key Laboratory of Novel Thin Film Solar Cells,ChineseAcademy of Science(KF200903).
中國科學院新型薄膜太陽能電池重點實驗室開放課題(KF200903)資助項目
O647;TM914.4