黃連-黃芩藥對化學成分的UPLC-PDA-MS 分析
2012-12-23 04:10:48張曉雷周明眉趙愛華茍小軍吳建兵惲祥惠邢麗娜
天然產(chǎn)物研究與開發(fā) 2012年11期
張曉雷,周明眉 ,趙愛華,茍小軍,吳建兵,惲祥惠,邢麗娜
1上海中醫(yī)藥大學;2 海綠谷制藥有限公司,上海201203;3 上海交通大學藥學院,上海200240
黃連、黃芩配伍使用出自《傷寒論》,《醫(yī)宗金鑒》名曰二黃湯。在中藥復方中出現(xiàn)頻率較高,如瀉心湯、黃連解毒湯、葛根芩連湯、普濟消毒飲等;2010版藥典中,黃芩黃連在20 多個復方中配伍使用。
中藥的配伍不等于簡單的藥物加和,不同的中藥配伍具有不同的藥效,其藥效物質(zhì)基礎(chǔ)也會有所不同。有關(guān)黃連黃芩藥對配伍的物質(zhì)基礎(chǔ)研究較少,其中李偉等[1]用毛細管電泳及液相色譜法研究黃連黃芩配伍過程化學成分的變化,發(fā)現(xiàn)黃連黃芩共煎產(chǎn)生沉淀,使有效成分的含量降低;齊粱[2]等應用聚酰胺柱層析、薄層層析和飛行時間質(zhì)譜對黃連黃芩藥對煎煮液的沉淀物進行了分析,沉淀物主要成分為藥材中的有效成分黃連堿、小檗堿、表小檗堿、巴馬亭、藥根堿及黃芩甙、漢黃芩甙等。盡管如此,但其研究內(nèi)容和方法有所不同。UPLC 是近年來新興的一種色譜技術(shù),它采用小顆粒填料色譜柱(粒徑小于2 μm)和超高壓系統(tǒng)(壓力大于105kPa)的新型液相色譜技術(shù),能顯著改善色譜峰的分離度和檢測靈敏度,同時大大縮短分析周期[3],因此特別適用于微量復雜混合物的分離和高通量研究。本文利用UPLC-PDA-MS 技術(shù)對黃連、黃芩、黃連黃芩藥對,以及含黃連黃芩的中成藥中的化學成分進行系統(tǒng)研究,建立全面反映黃連-黃芩藥對化學成分的分析方法,并進行定性及定量分析,最大限度的獲取相應的化學信息,從而為闡明黃連黃芩的配伍機制奠定基礎(chǔ)。
1 材料與儀器
1.1 實驗藥品及藥材
鹽酸藥根堿、鹽酸小檗堿、鹽酸巴馬汀、黃芩苷、漢芩黃苷、黃芩素、漢黃芩素及千層紙素A 購自上海融禾生物有限公司(純度>99%);黃連飲片(批號080201)產(chǎn)地四川,購于上海同濟堂;黃芩飲片(批號080123)產(chǎn)地河北,購自上海康橋中藥飲片有限公司。中成藥[三黃片(SHT)、一清顆粒(YQG)、牛黃上清丸(NHSQP)、蛤蚧定喘膠囊(GJDCC)及葛根芩連片(GGQLT)]購自上海養(yǎng)生堂。
1.2 實驗儀器
Waters 超高效液相色譜-串聯(lián)質(zhì)譜儀(ACQUITY/ZQ2000,美國WATERS 公司),配有Quity 二極管陣列檢測器(PDA)、電噴霧離子化源(ESI)、Masslynx 4. 1 工作站等;BP211D 型1/10 萬電子天平(Satorius Co.,Ltd,德國)等。
1.3 實驗試劑
甲醇(Fisher,美國)、甲酸(Tedia Company,美國)均為色譜純,其他試劑均為分析純。
2 方法與結(jié)果
2.1 色譜與質(zhì)譜條件
2.1.1 色譜條件
色譜柱:ACQUITY UPLC BEH C18柱(50 mm ×2.1 mm,1.7 μm);流動相:0.05%甲酸水溶液(A)和甲醇(B),梯度洗脫(0~2 min,88%→75% A;2~4 min,75%→69% A;4~6 min,69%→68% A;6~10 min,68%→35% A;10~11 min,35%→35%A;11~12 min,35%→88% A;12~14 min,88%→88% A);流速:0.35 mL/min;柱溫:30 ℃;進樣體積:3 μL;紫外檢測波長:280 nm。
2.1.2 質(zhì)譜條件
離子源:電噴霧離子源(ESI 源);檢測方式:正離子模式([M +H]+);掃描方式:一級全掃描;噴霧電壓:3.0 KV;錐孔電壓:50 V;霧化氣流量:500 L/h;脫溶劑溫度:350 ℃;離子源溫度:120 ℃。
2.2 對照品溶液的配制
2.2.1 對照品溶液的配制
分別精密稱取對照品鹽酸藥根堿、鹽酸小檗堿、鹽酸巴馬汀、黃芩苷、漢黃芩苷、黃芩素、漢黃芩素及千層紙素A 各約1000 μg,于10 mL 容量瓶中,加適量甲醇超聲溶解并稀釋至刻度線,得各對照品儲備液。取各對照品儲備液適量,分別稀釋,0.22 μm 微孔濾膜過濾,即得各對照品溶液。
2.2.2 混合對照品溶液的配制
分別精密稱取鹽酸藥根堿、鹽酸小檗堿、鹽酸巴馬汀、黃芩苷、漢芩黃苷、黃芩素、漢黃芩素及千層紙素A 各約1000 μg 于同一10 mL 的容量瓶中,加適量甲醇超聲溶解并稀釋至刻度線,得混合對照品儲備液(各成分濃度分別為136、99、93、92、117、110、106、115 μg/mL)。取混合對照品儲備液適量,稀釋,0.22 μm 微孔濾膜過濾,即得混合對照品溶液。
2.3 供試溶液的制備
2.3.1 提取物的制備
黃連、黃芩藥材分別粉碎至過20 目篩,干燥后分別稱取黃連50 g、黃芩50 g、黃連-黃芩藥對(1∶1)100 g,按1∶10(g∶mL)加水,浸泡30 min 后煎煮60 min,趁熱濾過,藥渣加8 倍量水再提取60 min,合并兩次提取液,靜置至室溫,3000 rpm 離心10 min,收集各上清液及黃連黃芩藥對(1∶1)沉淀,上清液70℃減壓濃縮,濃縮至稠膏;各稠膏及沉淀80 ℃真空干燥至恒重,稱重(平行三份,黃連平均提取率22.60%,黃芩平均提取率51.83%,黃連黃芩藥對平均提取率13. 38%,黃連黃芩藥對平均沉淀率27.52%),備用。
2.3.2 供試溶液的制備
2.3.2.1 藥材供試溶液的制備
分別稱取干燥藥材粉末:黃連50 mg、黃芩50 mg、黃連-黃芩藥對(1∶1)50 mg,于10 mL 帶塞玻璃離心管中,加65% 甲醇8 mL 稱重;室溫超聲60 min,稱重并用65%甲醇補足減失重量,3000 rpm 離心10 min,精密吸取上清液,適當稀釋后0.22 μm微孔濾膜過濾,即得各供試樣品溶液。
2.3.2.2 提取物供試溶液的制備
分別稱取干燥提取物粉末:黃連25 mg、黃芩25 mg、黃連-黃芩藥對(1∶1)25 mg 及藥對沉淀10 mg于10 mL 帶塞玻璃離心管中,其余操作步驟同上2.3.2.1。
2.3.2.3 含黃連黃芩藥對的中成藥供試溶液的制備
分別稱取干燥中成藥粉末:SHT 50 mg、YQG 50 mg、NHSQP 50 mg、GJDCC 50 mg 及GGQLT 50 mg 于10 mL 帶塞玻璃離心管中,其余操作步驟同上2.3.2.1。
2.4 方法專屬性考察及色譜峰的定性分析
2.4.1 色譜峰來源歸屬及專屬性考察
分別取上述配制的各對照品溶液、混合對照品溶液及供試樣品溶液,UPLC 自動進樣3 μL,使用優(yōu)化的梯度洗脫條件洗脫,確定各對照品、混合對照品及供試樣品中各成分的保留時間。色譜圖見圖1(I~X),由圖1 可知,黃連黃芩藥對提取液中共檢測到十二個主要的色譜峰。其中第1、2、3、4 和5 號峰均來自黃連,第6、7、8、9、10、11 和12 號峰均來自黃芩。這12 個主要的色譜峰保留時間各不相同,其他各峰互不干擾,證明該方法可行,專屬性較高,選擇性好。
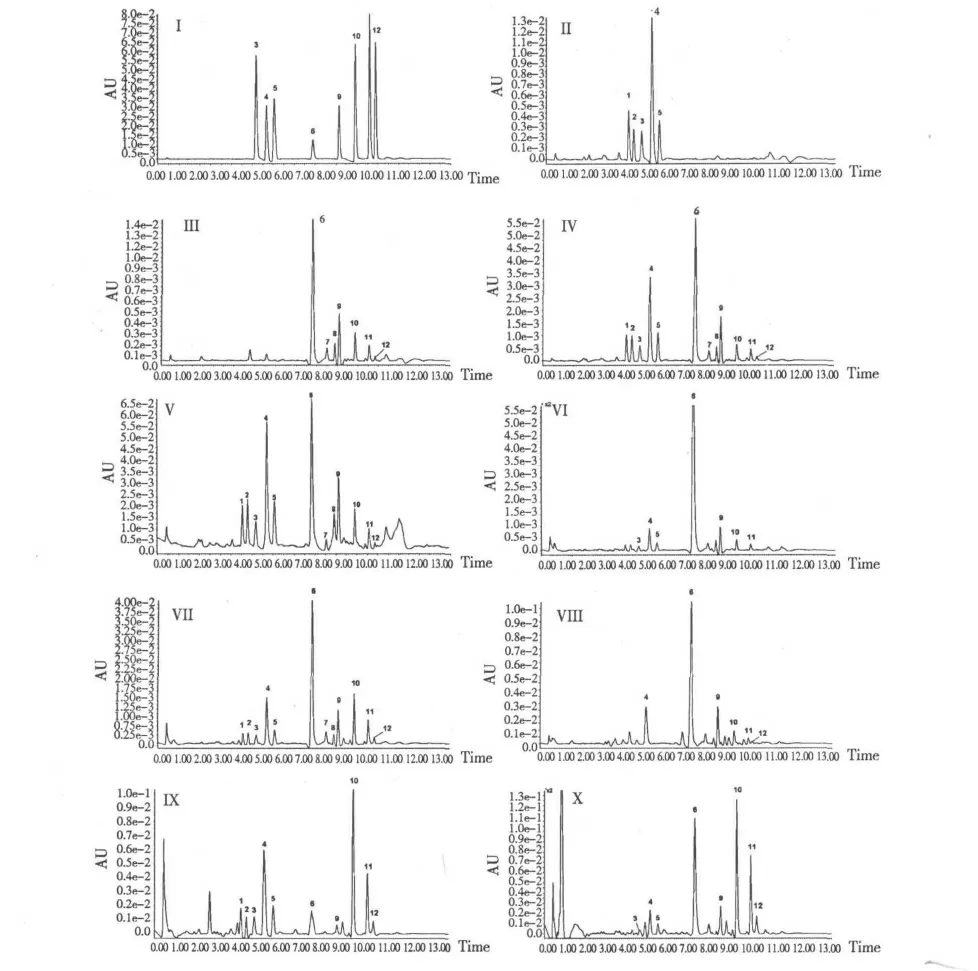
圖1 超高效液相色譜圖Fig.1 Typical UPLC chromatograms of the mixed
2.4.2 色譜峰的定性分析
通過比較供試樣品和對照品的色譜峰保留時間(tR),PDA 采集的色譜峰光譜和正離子方式檢測所得質(zhì)譜信息,確定峰3、4、5、6、9、10、11 和12 分別為藥根堿、小檗堿、巴馬汀、黃芩苷、漢黃芩苷、黃芩素、漢黃芩素和千層紙素A。峰2 的紫外光譜與峰4(小檗堿)相似(見圖2-II),質(zhì)譜圖顯示也極其相似(見圖2-I),且分子離子均為m/z 336,根據(jù)有關(guān)文獻[1,4],推斷為表小檗堿。峰1 的質(zhì)譜顯示分子離子為m/z 320(見圖2-I),并根據(jù)有關(guān)文獻[1,4],推斷為黃連堿。色譜峰1、2、3、4 及5 的質(zhì)譜信息,與文獻[4]報道相符。
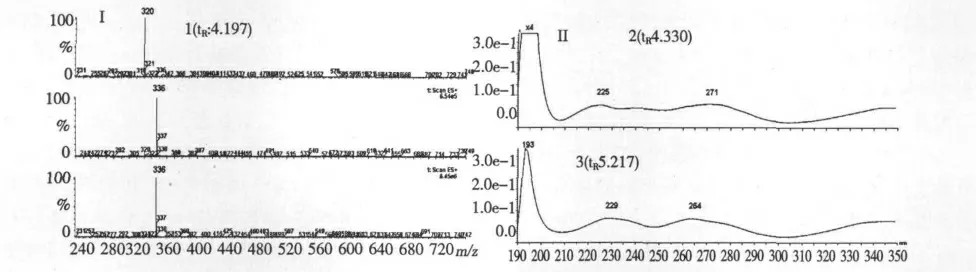
圖2 部分黃連成分(1、2、3)的質(zhì)譜圖(I);部分黃連成分(2、3)的紫外吸收圖(II)(3 小檗堿)Fig.2 Typical MS spectra of components 1,2,3 in Coptis (I);Representative UV spectra of components 2,3 in Coptis (II)(3.berberine)
2.5 線性關(guān)系的考察
精密量取上述配制的混合對照品儲備液適量,分別稀釋為10、30、100、300、1000、2000 倍等不同濃度的混合對照品系列溶液,過濾進樣,PDA 檢測,以8 種對照品的質(zhì)量濃度(x,μg/mL)為橫坐標,相應的峰面積為縱坐標(y),得回歸方程,見表1。
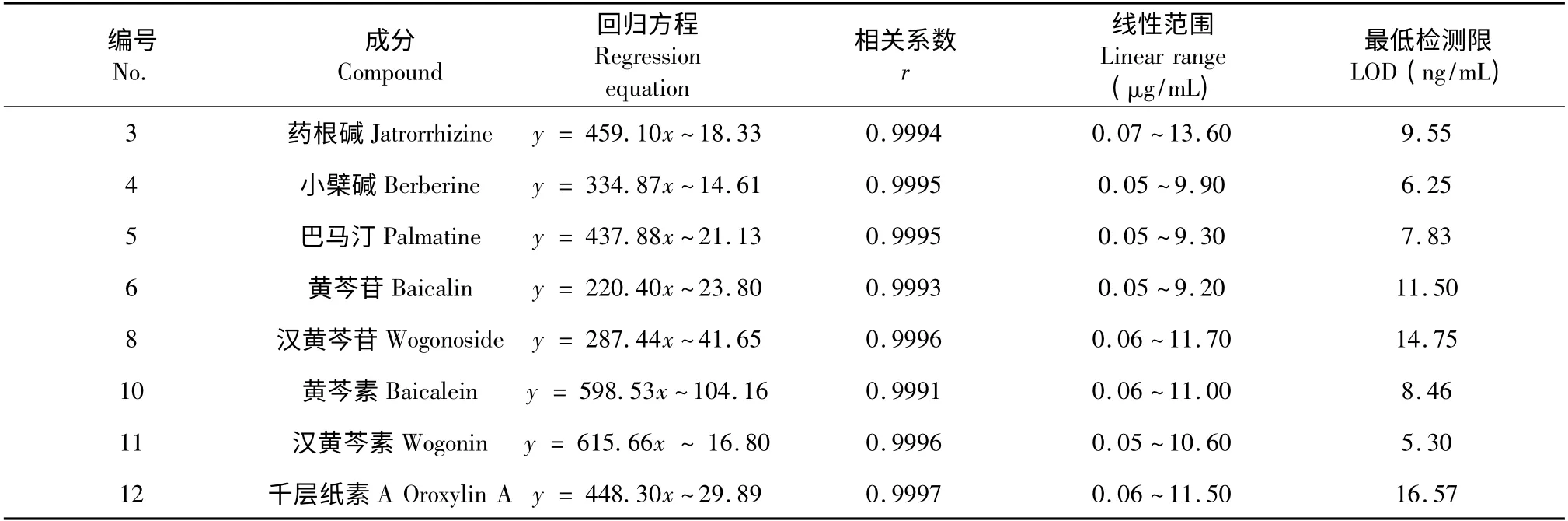
表1 各成分的線性回歸方程及LOD 測定結(jié)果Table 1 Linear regression equation and LOD data of the 8 identified compounds
2.6 精密度試驗
取“2.5”項下所配制的稀釋100 倍的混合對照品溶液,重復進樣6 次,PDA 檢測,計算各峰峰面積的RSD 值,結(jié)果分別為:鹽酸藥根堿0.10%、鹽酸小檗堿2.02%、鹽酸巴馬汀2.12%、黃芩苷0.45%、漢黃芩苷0.15%、黃芩素2.35%、漢黃芩素1.15%和千層紙素A 0.52%。說明精密度良好。
2.7 穩(wěn)定性試驗
取上述所配制的藥對提取物供試樣品溶液,分別在制備后的第0、3、6、12、16、24 小時進樣,PDA 檢測,計算各成分峰面積的RSD 值,結(jié)果分別為:藥根堿3.26%、小檗堿4.32%、巴馬汀4.63%、黃芩苷3.80%、漢黃芩苷4.92%、黃芩素4.61%、漢黃芩素4.47%和千層紙素A 3.78%。結(jié)果表明,8 個成分至少在24 h 內(nèi)穩(wěn)定。
2.8 重復性試驗
平行制備6 份藥對提取物供試樣品溶液,進樣,PDA 檢測,計算各成分峰面積的RSD 值,按所研究各成分的峰面積來考察重復性;結(jié)果RSD 值分別為:藥根堿3.59%、小檗堿2.01%、巴馬汀4.9%、黃芩苷1.13%、漢黃芩苷4.88%、黃芩素3.89%、漢黃芩素2.95%及千層紙素A 4.62%。結(jié)果表明:該方法重復性良好。
2.9 回收率試驗
精密稱取9 份黃連-黃芩藥對(1∶1)干燥藥材粉末,分別于10 mL 帶塞玻璃離心管中;每份10 mg,每3 份為1 組,各組分別加入混合對照品儲備液(各成分濃度分別為136、99、93、92、117、110、106、115 μg/mL)1、0.5、0.1 mL 做為高、中、低加樣組,然后再分別加65%甲醇至8 mL,稱重后按供試樣品溶液制備及測定方法進行操作,根據(jù)實際測得含量和已知含量的差值計算回收率。結(jié)果各成分高、中、低加樣回收率的平均值分別為:藥根堿101. 52%、101.88%、103. 33%;小檗堿99. 29%、103. 91%、100.97%;巴馬汀103. 76%、105. 47%、106. 77%;黃芩 苷102. 33%、94. 29%、99. 96%;漢 黃 芩 苷101.32%、99. 69%、96. 27%;黃 芩 素103. 75%、104.19%、105.27%;漢黃芩素104.53%、97.90%、99. 75%;千 層 紙 素 A 104. 18%、109. 09%、109.17%。各成分高、中、低加樣回收率的RSD 均小于5.0%。結(jié)果表明:該方法準確、可行。
2.10 樣品測定
取上述所配制的各供試樣品溶液,進樣,PDA檢測,根據(jù)標準曲線計算各成分含量。按藥根堿、小檗堿、巴馬汀、黃芩苷、漢黃芩苷、黃芩素、漢黃芩素及千層紙素A 的含量來研究藥對黃連、黃芩配伍的相互作用;此外,利用所建立的方法成功地對含該藥對的部分中成藥進行了質(zhì)量考察,其結(jié)果見表2。實驗發(fā)現(xiàn),黃連配伍黃芩后,產(chǎn)生大量黃色絮狀沉淀,使黃連黃芩提取物重量降低26%;對藥對提取物及沉淀進行分析與測定,發(fā)現(xiàn)提取物中含藥根堿0. 75%、小檗堿0. 45%、巴馬汀2. 03%、黃芩苷8.56%、漢黃芩苷3.89%、黃芩素0.51%,而沉淀中含藥根堿1.08%、小檗堿17.45%、巴馬汀1.12%、黃芩苷37.31%、漢黃芩苷4.04%、黃芩素0.81%,均高于提取物,其中小檗堿、黃芩苷最多。
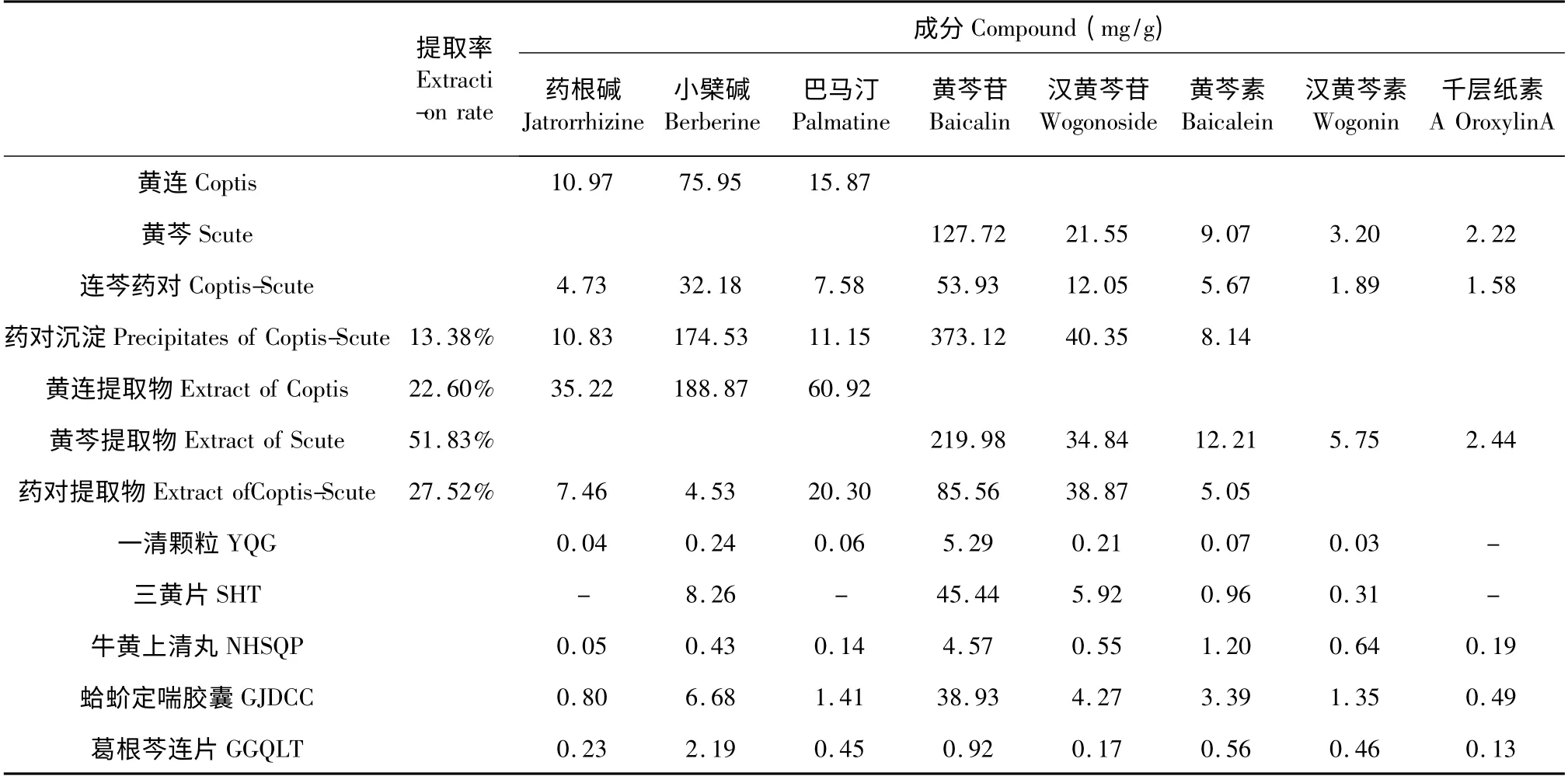
表2 樣品測定結(jié)果(平均結(jié)果,mg/g,n = 3)Table 2 Quantification results of samples (All data expressed as mean,mg/g,n = 3)
3 小結(jié)與討論
本實驗在同一條件下,對黃連、黃芩及黃連黃芩藥中的化學成分進行系統(tǒng)研究,建立全面反映黃連、黃芩及黃連黃芩藥對所含化學成分的分析方法,效果較好,方法簡單、方便、可行,并與有關(guān)文獻[1,5]相比,也大大的縮短了分析時間;應用該方法對黃連、黃芩及黃連黃芩藥對進行定性及定量分析,最大限度的獲取相應的化學信息,從而為闡明黃連黃芩的配伍機制奠定基礎(chǔ);并且還成功地對部分含該藥對的中成藥進行了含量測定。本實驗可以定性并能定量的成分較多,雖然只測定其中8 種成分,但可為中藥及中藥制劑的質(zhì)量標準制定提供了一個較為合理的思路;目前,中藥及中藥制劑的質(zhì)量標準制定,往往以其中的一種或兩種成分為指標進行質(zhì)量控制,但中藥的療效往往是多種中藥或多種成分的協(xié)同作用,所以以其中的一兩種成分作為質(zhì)量控制指標缺乏科學性,若盡可能多的檢測相關(guān)成分,則可顯著提高中藥或中藥制劑的質(zhì)量標準,為其安全有效可控提供可靠保障。
實驗中發(fā)現(xiàn)藥對混煎時,提取物重量降低近30%,同時也發(fā)現(xiàn)沉淀中主要含有與煎液成分相同的化學成分,即藥根堿、小檗堿、巴馬汀、黃芩苷、漢黃芩苷、黃芩素、及漢黃芩素等,與有關(guān)文獻[1,2]一致。沉淀中小檗堿、黃芩苷分別多達17. 45%、37.31%,遠遠高于提取物中的相應含量0. 45%、8.56%。此外大量研究證實,產(chǎn)生沉淀的原因是由于黃連中的原小檗堿型生物堿為堿性化合物,與黃芩中的多酚類化合物黃芩苷等酸性化合物相互結(jié)合,生成分子量較大的水不溶物而產(chǎn)生沉淀,同時藥理實驗也證明沉淀物具有藥效作用[2]。根據(jù)中醫(yī)理論,黃連黃芩藥對屬清熱燥濕配伍的經(jīng)典藥對,二者皆屬苦寒之品,均能清熱燥濕、瀉火解毒,常相須合用起協(xié)同作用,以發(fā)揮最佳療效;由于中藥方劑傳統(tǒng)入藥方式為湯劑,其配伍共煎后產(chǎn)生的沉淀大部分以混濁液的形式一起服用,而沉淀一般是兩類成分形成的復合物,進入人體內(nèi),受環(huán)境影響不斷解離,起到緩釋的作用,對臨床療效的影響不大,而對于相關(guān)的成藥或有關(guān)研究,由于制備工藝中的過濾步驟除去了大部分沉淀,勢必對其療效造成較大影響;在中醫(yī)處方中常見對特殊藥味的“先煎”、“后下”、“另燉”等醫(yī)囑,其實都是前人通過實踐摸索出各藥味的性質(zhì)后,為避免其有效成分的損失或增加煎出量而采取的相應措施。因此,在明確了含酸堿成分對藥的方劑煎煮會導致酸、堿性有效成分煎出量減少這樣的事實,就應該采用分煎方式,盡量避免有效成分的損失,以保證有效成分最大限度的保留;傳統(tǒng)的湯劑人藥也可以考慮“另煎”(即分煎)的方式,以提高療效,降低費用。
本實驗采用藥材和水1∶10 的提取溶劑比例,目的考察其習慣提取方法的提取效率及藥對配伍在提取過程中的相互影響,此外根據(jù)黃連、黃芩有效成分的溶解性(黃芩苷溶解度甲醇1569 μg/mL、乙醇1458 μg/mL、水60 μg/mL[6])及有關(guān)文獻[7],分別進行溶媒甲醇及65%甲醇水提取預試驗,結(jié)果發(fā)現(xiàn)采用65%甲醇提取各有效成分效果較好,故采用65%甲醇提取并進行相應的定性定量分析,結(jié)果取得滿意效果。
1 Li W(李偉),Song FR(宋鳳瑞),Liu ZQ(劉志強),et al.Studies on change of chemical composition in Coptis-Scute herb couple by using HPCE and HPLC.Acta Pharm Sin(藥學學報),2008,43:191-194.
2 Qi L(喬梁),Peng JR(彭嘉柔),Pu XS(濮訓生),et al.Analysis of the precipitates in decoction of Coptis-Scute herb couple.China J Chin Mater Med(中國中藥雜志),1999,24:352-353.
3 Waters Corporation.UPLC TM:Separation Science Redefined.2006,720000880.
4 Deng YT(鄧雅婷),Liao QF(廖瓊峰),Bi KS(畢開順),et al.Analysis of the main components of Coptis-Evodia herb couple by HPLC-DAD-MS. Acta Pharm Sin(藥學學報),2008,43:299-302.
5 Shi R(石榮),Ma YM(馬越鳴),Zhang N(張寧),et al.Study on contents of alkaloids and flavonoids in Xiexin decoction with different combinations. J Chin Pharm(中國藥學雜志),2007,42:1771-1773.
6 Wang H(王弘),Chen JM(陳濟民),Zhang QM(張清民).Determination of the physical chemistry constants of Baicalin.J Shenyang Pharm Univ(沈陽藥科大學學報),2003,17:105-106.
7 Huang YB,Wu PC,Su CS,et al.Simultaneous quantification of twelve bioactive components in San-huang-xie-xin-tang by HPLC.Phytochem Anal,2006,17:439-446.
