磷脂在膜結構間的交換:溫度和離子強度的影響*
2013-04-14 06:21:38蔣中英張國梁馬晶朱濤
物理學報 2013年1期
蔣中英 張國梁馬晶朱濤
1)(伊犁師范學院電子與信息工程學院,伊寧 835000)
2)(南京大學物理學院,南京 210093)
(2012年2月29日收到;2012年8月6日收到修改稿)
1 引言
生物界中存在著數千種天然磷脂.在不同生物膜中,各類磷脂組分比例的差異性直接與特異性生物膜功能相關.為建立并維持膜內磷脂組分比例,磷脂需要在不同的生物膜結構間實現跨膜運輸與交換[1].因此,膜間磷脂交換是一項重要的生理活動.同時,藥物輸運也涉及磷脂跨膜交換問題.許多以脂質體為載體的藥物正處于臨床應用或審核階段,這些藥物的能效與磷脂跨膜交換存在關聯.例如,在鼠類卵巢細胞的原位研究中,正電性磷脂包裹的多孔硅小球被報道具有較高的藥物輸送能力[2];在鼠、兔活體研究中,正電性磷脂載運的DNA,siRNA被證實具有顯著的細胞轉染表達效能[3],而具有生物活性的細胞膜顯負電性.因此,異電性磷脂膜間的接觸和磷脂交換機理及其影響因素特別值得深入認知.
正、負電性磷脂膜間的平衡磷脂交換量與磷脂跨膜交換速率已經被深入研究.MacDonald等[4],Saeki等[5]發現異電性膜組分分子間的靜電吸引作用是磷脂跨膜交換的主要驅動力.帶電磷脂組分比例越高,平衡磷脂交換量越大.文獻[6—9]考察了磷脂跨膜交換速率的多種影響因素,膜間磷脂交換隨著帶電磷脂組分比例、囊泡濃度、磷脂疏水性、膜組分側向擴散系數、膜曲率的提高而增快.這些研究為深入理解磷脂跨膜交換過程提供了參考.然而不同于平衡磷脂交換量與磷脂跨膜交換速率,交換過程中磷脂膜間接觸狀態(包括總磷脂膜接觸面積等)的研究卻仍處于起步階段.通過冷凍電鏡與濁度儀測試,Ichikawa等[5],Lehn等[10]觀測到伴隨著磷脂跨膜交換的異電性囊泡聚集行為.但是由于懸浮囊泡表征手段的限制,實時、定量的囊泡聚集態研究仍是空白.界面膜體系(包括支撐膜、吸附囊泡等)具有便于表征的優點.平面表征手段如原子力顯微鏡(AFM)、表面等離子體共振(SPR)和石英電子微天平及耗散系數測量儀(QCM-D)等都能用于觀測界面膜體系的動態變化過程.在先前的研究中,我們即采用吸附囊泡與支撐膜體系,實現了對磷脂跨膜交換過程中磷脂膜間接觸狀態的半定量表征,并初步探索了膜帶電量、膜流動性、膜曲率與磷脂膜間最大總接觸面積的關系[9].
在本研究中,我們進一步采用QCM-D研究了溫度和離子強度對正電性支撐膜與負電性囊泡磷脂交換過程中膜間接觸面積的影響.QCM-D是一種高精度、實時記錄界面吸附層質量與黏彈性變化的表征技術,被廣泛應用于測量高分子聚合物和蛋白質的動力學吸附過程.如Decher等[11],Kasemo等[12]利用QCM-D研究了溫度、鹽濃度、pH值等對SiO2表面吸附囊泡向支撐膜轉變的臨界覆蓋率的影響.在此,我們利用QCM-D手段表征了支撐膜表面異電性囊泡的吸附量,從而間接獲取膜間接觸狀況的信息.研究表明:溫度和鹽濃度的提高降低最大膜間總接觸面積.囊泡吸附速率和磷脂跨膜交換速率的變化在其中發揮著關鍵的調節作用.本研究有助于加深理解部分磷脂跨膜交換參與的生物學現象,并對脂質體藥物載體的設計與應用提供參考.
2 實驗部分
2.1 材料
二油酰基磷脂酰膽堿(DOPC)、二油酰基磷脂酰絲氨酸(DOPS)與二油酰基三甲氨基丙烷(DOTAP)購于 Avanti Polar Lipids公司.DOPC,DOTAP,DOPS分別為雙電性、正電性與負電性磷脂(分子式如圖1所示).所有磷脂跨膜交換實驗都是在三(羥甲基)氨基甲烷鹽酸鹽-鹽酸(Tris-HCl)緩沖液中進行的.緩沖液中Tris的濃度為100mmol/L,pH值為8,離子強度由NaCl濃度(CNaCl)調節.
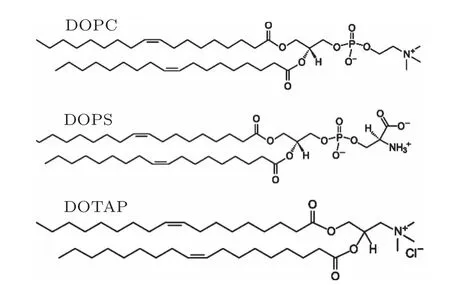
圖 1 DOPC,DOTAP和 DOPS的分子式 DOPC,DOTAP,DOPS分別為電雙性、正電性與負電性磷脂
2.2 囊泡制備
囊泡通過擠出法制備[13],其過程簡述如下:氮氣干燥磷脂氯仿溶液形成磷脂膜,進一步通過真空干燥箱干燥;緩沖液水化磷脂膜,形成多層脂質體;通過提取器(100 nm孔徑濾膜)循環擠壓脂質體溶液50次制備單層磷脂囊泡;制備的囊泡通過粒度儀(NanSight公司LM10納米粒度分析儀)與冷凍電鏡(FEI公司Tecnai 200 kV冷凍電鏡)方法檢測粒徑,其流體力學半徑(Rh)為56 nm±5 nm(圖2).
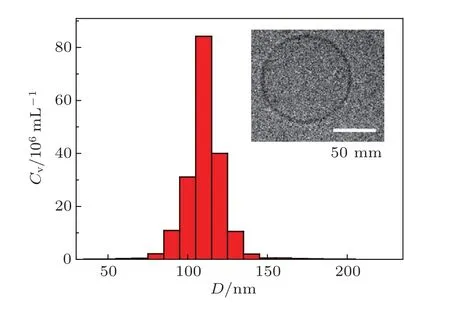
圖2 囊泡粒徑的動態光散射和冷凍電鏡測試,其中D為囊泡的流體力學直徑,Cv是囊泡的濃度
2.3 石英電子微天平及耗散系數測量儀測試
QCM-D高精度地實時記錄界面吸附層的質量與黏彈性變化信息,其測量參數包括共振頻率偏移值(Δf)和能量耗散系數偏移值(ΔD).Δf與吸附層質量(Δm)近似符合Sauerbrey關系[14]:
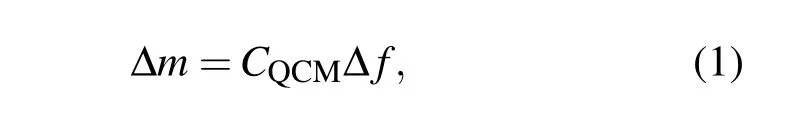
其中CQCM是質量感應常數,ΔD與吸附層的黏彈性質相關.如果吸附物質的力學性質趨近于液體性質(高黏度、高耗散性,如磷脂囊泡),則產生較高的ΔD(飽和吸附囊泡層的ΔD大于10-6);如果吸附物質的力學性質趨近于固體性質(高彈性模量、低耗散性,如磷脂支撐膜),則產生較低的ΔD(飽和吸附磷脂支撐膜的ΔD約10-7)[12].
磷脂跨膜交換實驗使用了SiO2涂層的石英芯片(基頻為4.95 MHz),在E1實驗平臺(瑞典QSense公司)上進行.溶液的進樣流速被控制在20μL/min.具體步驟如下:首先在緩沖液環境下建立穩定的基線;然后通過囊泡融合在SiO2表面形成正電性磷脂支撐膜結構;艙室內剩余的囊泡通過緩沖液漂洗除去;進一步通入負電性磷脂囊泡,磷脂在負電性囊泡與正電性支撐膜間進行跨膜交換;磷脂交換結束后,再次通入緩沖液;每個交換實驗重復3次得到平均值與標準方差.文中所有數據的諧頻數為7.
3 結果
負電性囊泡與正電性支撐膜間的磷脂跨膜交換實驗通過QCM-D表征.圖3和圖4展示了不同溫度、離子強度條件下測得的Δf-t與ΔD-t曲線.所有曲線以t=35 min為界可分為兩個部分:前半部分時間對應著正電性囊泡在SiO2表面融合形成支撐膜結構(成膜過程);后半部分時間對應著負電性囊泡與正電性支撐膜進行磷脂跨膜交換(交換過程).
3.1 成膜過程
對于DOTAP/DOPC組分比例為20/80的正電性囊泡的成膜過程,首先-Δf與ΔD隨時間增大,表示囊泡逐漸吸附到SiO2表面.隨后-Δf與ΔD降低,表示在達到囊泡臨界表面覆蓋率后,吸附囊泡開始顯著破裂形成片層支撐膜結構.最后Δf穩定在-24.2 Hz±0.5 Hz,ΔD平衡在(5.0±1.2)×10-7,說明形成了完整的支撐膜結構.對于DOTAP/DOPC組分比例為50/50的正電性囊泡的成膜過程,-Δf與ΔD單調增加并最終平衡在-22.0 Hz±0.7 Hz與(6.4±3.4)×10-7,同樣表明形成了完整的支撐膜結構.以上兩種組分磷脂囊泡的成膜過程分別對應著兩條成膜路徑:前者,囊泡所帶正電量較低(0.09 C/m2),與SiO2襯底(負電性)吸引作用較弱,需要借助吸附囊泡之間的壓力(即通過達到囊泡臨界表面覆蓋率)實現成膜;后者,囊泡所帶正電量較高(0.23 C/m2),與SiO2襯底吸引作用較強,在底面吸附后直接破裂成膜.Brisson等[15]通過AFM與橢圓偏振光譜測試也觀察到了這種襯底-囊泡相互作用強弱引起的不同的囊泡成膜途徑.
3.2 交換過程
在不同溫度條件下,交換過程測得的Δf與ΔD隨t的變化趨勢符合相同的規律(圖3).首先-Δf與ΔD隨時間升高,表示負電性囊泡開始吸附到正電性支撐膜表面(吸附主導階段).隨后-Δf與ΔD達到極值-Δfmin與ΔDmax.其中-Δfmin對應著交換過程中支撐膜表面的最大囊泡吸附量[9].之后-Δf與ΔD開始降低,表示隨著磷脂跨膜交換,囊泡與支撐膜間的靜電吸引作用逐漸喪失,囊泡在支撐膜表面的解吸附主導了襯底的質量與黏彈性變化(解吸附主導階段).最后Δf與ΔD分別穩定在-23.5 Hz±2.1 Hz與 (1.2±0.6)×10-6,緩沖液漂洗不會顯著改變平衡的Δf與ΔD值,說明大多數吸附囊泡實現了解吸附[16].這個磷脂跨膜交換過程在Kasemo與我們之前的研究中已被詳細闡述[9].從Δf開始下降到最終Δf恢復平衡之間的時間被定義為tint.它表示囊泡與異電性支撐膜的總接觸作用時間.在時間tint內,磷脂在囊泡與支撐膜間進行跨膜交換.如果平衡磷脂交換量確定,則tint越大,磷脂跨膜交換速率越低;反之亦然.由圖3可見,tint隨著溫度的升高而降低.溫度每升高10°C,tint下降14%—38%.由于溫度變化不會顯著改變磷脂跨膜交換的靜電作用驅動力和平衡磷脂交換量[4],所以溫度的升高加速了磷脂跨膜交換過程.同時,由圖3可見,Δfmin隨著溫度的升高而降低.溫度每升高10°C,-Δfmin降低24%—60%.
在不同離子強度條件下,交換過程測得的Δf與ΔD隨t的變化趨勢具有顯著差別(圖4).當氯化鈉濃度(CNaCl)為400 mmol(離子強度約為 0.4mol/L),支撐膜 DOTAP/DOPC組分比為20/80,囊泡內DOPS/DOPC組分比任意時,Δf隨著時間無顯著變化,表示支撐膜表面無囊泡吸附;當CNaCl為400 mmol,支撐膜DOTAP/DOPC組分比為50/50,囊泡內DOPS/DOPC組分比為20/80時,Δf隨著t單調減小并最終穩定在-8.3 Hz±1.5 Hz,ΔD單調增加并最終平衡在(3.6±0.4)×10-6,表示囊泡不可逆吸附在支撐膜表面.出現以上兩種情況的原因可能是溶液中的離子屏蔽了囊泡與支撐膜間的靜電吸引作用,導致囊泡無法在支撐膜表面吸附或吸附后沒有進行有效的磷脂跨膜交換(致使囊泡無法進一步解吸附).對于CNaCl為400 mmol,支撐膜DOTAP/DOPC組分比50/50,囊泡DOPS/DOPC組分比50/50和其他鹽濃度(CNaCl為0 mmol或100 mmol,離子強度分別約為10-8mol/L和0.1 mol/L)條件下的實驗,-Δf與ΔD隨時間先增加后減小,表示經歷了囊泡吸附主導與解吸附主導的磷脂交換的兩個階段.同時,tint隨著鹽濃度的提高而延長,CNaCl提高 100 mmol,tint增加 21%—148%.因為鹽濃度升高屏蔽了磷脂跨膜交換的靜電作用驅動力,降低平衡磷脂交換量[4],所以離子強度的提高降低了磷脂跨膜交換速率.由圖4中還可以得到,-Δfmin隨著離子強度的提高而降低,CNaCl提高100 mmol,Δfmin降低6%—39%.
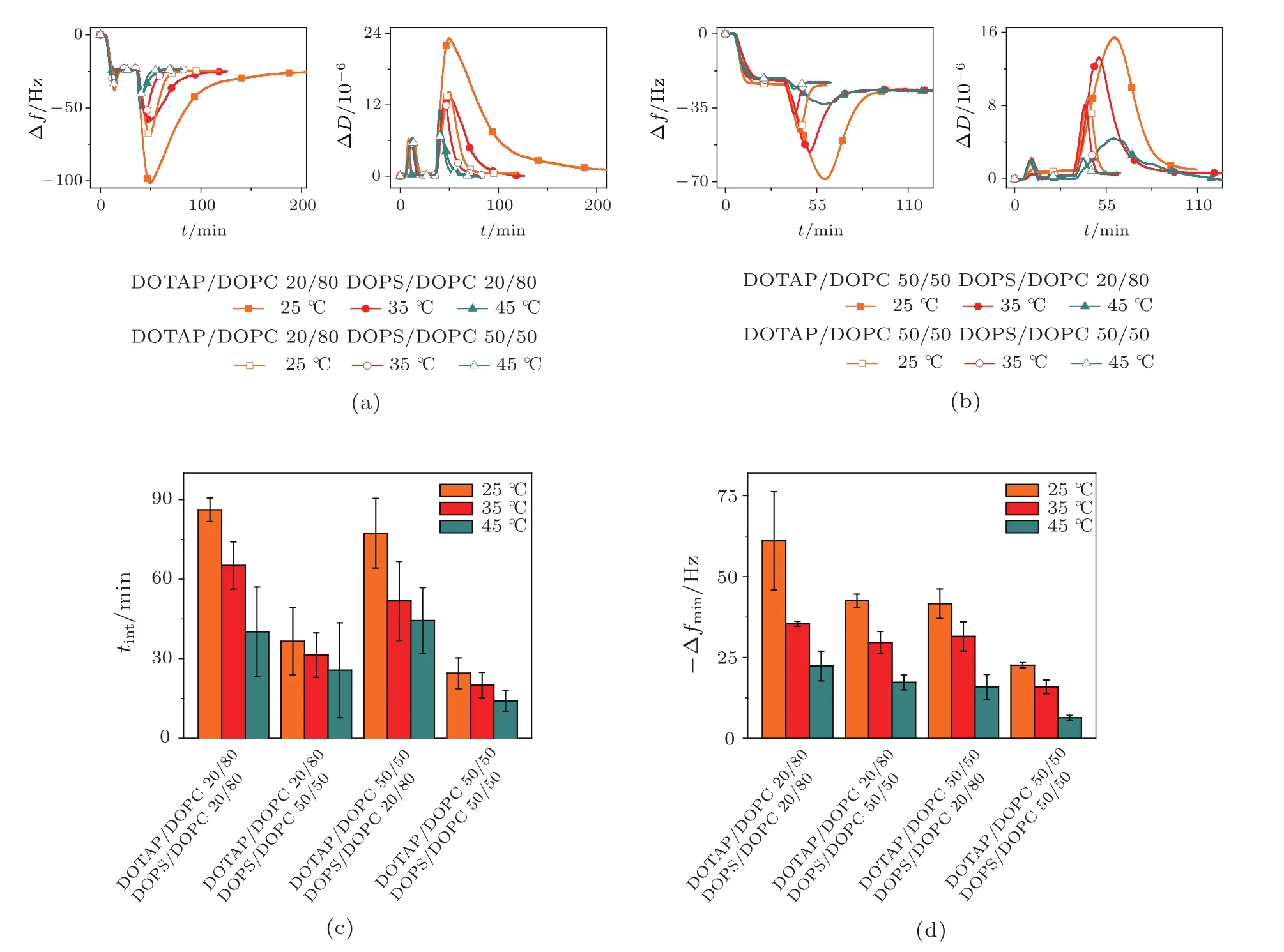
圖3 溫度對磷脂跨膜交換的影響 (a),(b)Δf-t與ΔD-t曲線;(c),(d)在不同溫度條件下測得的tint與-Δfmin統計數據直方圖;實驗所處溫度被標注在各圖中,該組實驗Tris-HCl緩沖液中CNaCl為100 mmol
4 討論
單個囊泡和支撐膜之間的接觸面積與囊泡在支撐膜表面吸附產生的形變(囊泡的高寬比變化)直接相關.QCM-D的-ΔD/Δf(表示單位質量吸附層所對應的黏彈性)被證實能夠用于表征吸附囊泡的形變情況[12].囊泡形變程度越高(高寬比越低于1),產生的-ΔD/Δf越低.不同溶液環境條件下交換過程的ΔD-Δf曲線需仔細地分析.如圖5所示,對于能夠有效進行磷脂跨膜交換的實驗,ΔD-Δf曲線構成一個環路,曲線的下半段對應著囊泡吸附主導的階段,上半段對應著囊泡解吸附主導的階段.吸附主導階段的-ΔD/Δf值一般略小于解吸附主導階段的-ΔD/Δf值,表明隨著磷脂跨膜交換的進行,囊泡與支撐膜間的靜電吸引作用逐漸減弱,吸附囊泡的形變程度略微降低.以時間軸統計-ΔD/Δf值,發現單條實驗曲線的-ΔD/Δf標準方差與平均值之比都低于12%,說明在整個交換時間段中,吸附囊泡的形變差異性并不非常顯著.統計所有實驗曲線的平均-ΔD/Δf值得-ΔD/Δf=0.36±0.03,說明不同溶液環境帶來的吸附囊泡形變差異性也較小.因此,在以下的囊泡-支撐膜總接觸面積研究中,單個囊泡與支撐膜的接觸面積被近似為相同.
由于單個吸附囊泡與支撐膜的接觸面積相對恒定,所以囊泡與支撐膜的總接觸面積直接取決于囊泡的吸附量,即QCM-D實驗參數Δfmin在代表支撐膜表面囊泡吸附最大量的同時,也對應著最大的囊泡-支撐膜總接觸面積.我們之前的研究發現,囊泡在異電性支撐膜表面的最大吸附量是由囊泡吸附速率和磷脂跨膜交速率之間的競爭決定的[9].囊泡吸附速率的提高有利于增加最大囊泡吸附量,而磷脂跨膜交換速率的提高加速了囊泡與支撐膜間靜電吸引作用的喪失和囊泡的解吸附過程,不利于增加最大囊泡吸附量.
圖3中的QCM-D數據顯示,Δfmin隨著溫度的升高而降低,表示交換過程中囊泡的最大吸附量和最大囊泡-支撐膜總接觸面積隨著溫度的升高而降低;同時,tint隨著溫度的升高而降低,即磷脂跨膜交換速率隨著溫度的升高而增加.基于囊泡吸附速率和磷脂跨膜交換速率對于最大囊泡吸附量的競爭效應,溫度的提高對磷脂跨膜交換的加速效應應強于對囊泡吸附的加速效應.這可以通過以下的分析簡單地理解.一方面,由于囊泡的吸附過程是受囊泡擴散速率限制的[17],所以溫度的升高會增加囊泡的擴散和吸附速率.根據Stokes-Einstein方程[9]:
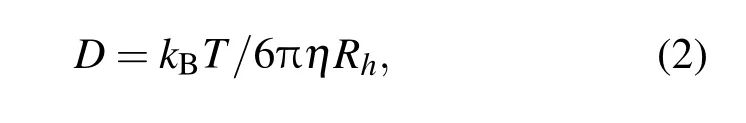
其中D是囊泡擴散速率,kB是波爾茲曼常數,T是絕對溫度,η是溶液黏度.因此,實驗溫度提升10—20°C,囊泡擴散與吸附速率增加3%—6%.另一方面,半融合結構是磷脂在異電性磷脂膜間交換所依賴的中間態結構(如圖6所示)[18].在半融合結構中,磷脂通過側向擴散實現跨膜交換[18].溫度的提高顯著增加磷脂的側向擴散速率.Papahadjopoulos等[19]研究表明,溫度從20°C提升到40°C,液晶相磷脂膜的側向擴散速率提升一個量級.隨著磷脂側向擴散速率的增大,磷脂跨膜交換速率也被顯著提高.因此,溫度的升高增加囊泡吸附速率和磷脂跨膜交換速率,但對后者的增強效應強于前者,導致最大囊泡吸附量和最大囊泡-支撐膜總接觸面積降低.
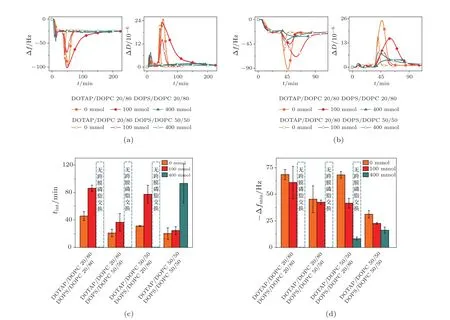
圖4 離子強度對磷脂跨膜交換的影響 (a),(b)Δf-t與ΔD-t曲線;(c),(d)在不同鹽濃度(離子強度)條件下測得的tint與-Δfmin統計數據直方圖;實驗所用CNaCl被標注在各圖中,該組實驗的實驗溫度為25°C
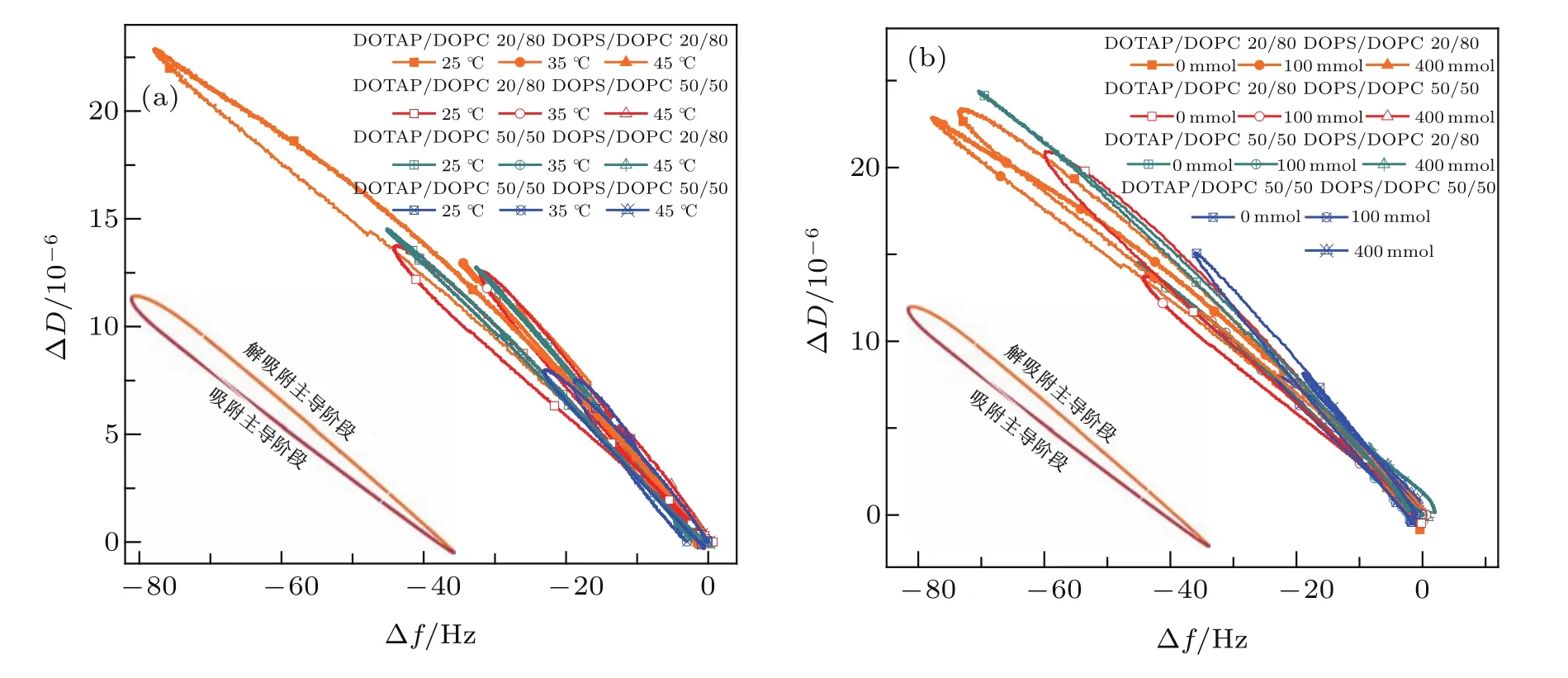
圖5 交換過程的囊泡在異電性支撐膜表面吸附產生的ΔD-Δf曲線(交換過程前的ΔD,Δf值被歸零) (a),(b)的數據分別取自圖3和圖4;吸附、解吸附主導階段對應曲線示意圖標明在圖中的左下角
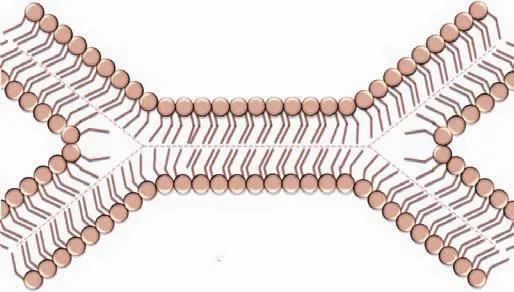
圖6 交換過程中半融合結構示意圖 兩磷脂膜中相鄰兩葉發生融合,較遠側兩葉保持完整,在半融合結構中,磷脂通過側向擴散實現跨膜交換
圖4中的QCM-D數據顯示,Δfmin隨著離子強度的升高而降低,表示交換過程中囊泡的最大吸附量和最大囊泡-支撐膜總接觸面積隨著離子強度的增大而降低;同時,tint隨著離子強度的升高而增大,即磷脂跨膜交換速率隨著離子強度的升高而降低.基于囊泡吸附速率和磷脂跨膜交換速率對于最大囊泡吸附量的競爭效應,離子強度的提高對磷脂跨膜交換的減速效應應弱于對囊泡吸附的減速效應.這也可以通過以下的分析簡單的理解:一方面,囊泡與支撐膜間的靜電吸引作用是囊泡向支撐膜靠近的主要驅動力,而長程的靜電相互作用被離子以雙電層形式強烈屏蔽,CNaCl為0,100,400 mmol緩沖溶液的Deybe半徑 (電場屏蔽尺度)分別為 3.1μm,1 nm,0.5 nm,因此,離子強度的增加顯著降低囊泡的吸附速率;另一方面,鹽離子破壞磷脂膜表面的有序水化層結構,促進短程的磷脂膜間接觸[8,20],有利于加速實現磷脂的跨膜交換,緩解長程靜電相互作用減弱帶來的磷脂跨膜交換速率降低.因此,離子強度的升高降低了囊泡吸附速率和磷脂跨膜交換速率,但對于前者的減弱效應強于后者,導致最大囊泡吸附量和最大囊泡-支撐膜總接觸面積降低.
5 結論
磷脂在異電性膜結構間的交換是一個復雜的過程.本文采用界面膜體系通過QCM-D表征手段研究了溫度和離子強度對該過程的影響.研究表明:1)溫度的升高加速囊泡與支撐膜間磷脂跨膜交換過程,降低囊泡的最大吸附量;離子強度的升高減緩囊泡與支撐膜間磷脂跨膜交換過程,降低囊泡最大吸附量;2)在各溶液條件下,支撐膜表面吸附囊泡形變的差異性較低,因此囊泡的吸附量直接對應著囊泡與支撐膜的總接觸面積;3)溫度升高對磷脂跨膜交換的加速效應強于對囊泡吸附的加速效應,造成囊泡-支撐膜最大總接觸面積隨溫度升高而降低;離子強度升高對磷脂跨膜交換的減速效應弱于對囊泡吸附的減速效應,造成囊泡-支撐膜最大總接觸面積隨離子強度升高而降低.
許多磷脂跨膜交換過程的問題仍待解決,如膜間接觸面積的動力學變化過程、磷脂包裹顆粒與囊泡的交換差異性[21]、磷脂囊泡脫離支撐膜表面的方式[22]等.實際的醫藥學領域應用還要涉及囊泡在支撐膜表面的破裂和內含物釋放問題[23].進一步探索膜結構間的接觸信息將有助于加深我們對以上問題的理解.
[1]Holthuis J C M,Levine T P 2005Nat.Rev.Mol.Cell Biol.6 209
[2]Liu J,Jiang X,Ashley C,Brinker C J 2009JACS131 7567
[3]Zuhorn I S,Engberts J B F N,Hoekstra D 2007Eur.Biophys.J.36 349
[4] Pantazatos D P,Pantazatos S P,MacDonald R C 2003J.Membr.Biol.194 129
[5]Saeki D,Sugiura S,Baba T,Kanamori T,Sato S,Mukataka S,Ichikawa S 2008J.Colloid Interface Sci.320 611
[6]Reinl H M,Bayerl T M 1994Biochemistry33 14091
[7]Jones J D,Thompson T E 1989Biochemistry28 129
[8]Stamatatos L,Leventis R,Zuckermann M J,Silvius J R 1988Biochemistry27 3917
[9]Zhu T,Jiang Z,Ma Y 2012Colloids Surf.B 97 155
[10]MarchiArtzner V,Jullien L,Belloni L,Raison D,Lacombe L,Lehn J M 1996J.Phys.Chem.100 13844
[11]Seantier B,Breffa C,Felix O,Decher G 2005J.Phys.Chem.B 109 21755
[12]Reimhult E,Hook F,Kasemo B 2003Langmuir19 1681
[13]Zhu T,Xu F,Yuan B,Ren C,Jiang Z,Ma Y 2012Colloids Surf.B 89 228
[14]Sauerbrey G 1959Z.Angew.Phys.155 206
[15]Richter R P,Brisson A R 2005Biophys.J.88 3422
[16]Wikstrom A,Svedhem S,Sivignon M,Kasemo B 2008J.Phys.Chem.B 112 14069
[17]Keller C A,Glasmastar K,Zhdanov V P,Kasemo B 2000Phys.Rev.Lett.84 5443
[18]Lei G H,MacDonald R C 2003Biophys.J.85 1585
[19]Wu E S,Jacobson K,Papahadjopoulos D 1977Biochemistry16 3936
[20]Sapuri A R,Baksh M M,Groves J T 2003Langmuir19 1606
[21]Ding H M,Tian W D,Ma Y Q 2012ACS Nano6 1230
[22]Li J B,Zhang Y,Yan L L 2001Angew.Chem.Int.Edit.40 891
[23]An Z H,Tao C,Lu G,Mohwald H,Zheng S P,Cui Y,Li J B 2005Chem.Mater.17 2514?
