淋巴瘤樣肉芽腫病1例
2013-04-18 09:05:24宋美君吳宏成潘登林志輝
浙江醫學 2013年3期
宋美君 吳宏成 潘登 林志輝
淋巴瘤樣肉芽腫病1例
宋美君 吳宏成 潘登 林志輝
患者男,66歲,農民。因“咳嗽、咳痰2月余,加重1周”于2010年4月8日入院。患者咳嗽呈陣發性,不劇,痰色白,量少,無發熱、咯血、盜汗、消瘦、氣促、紫紺等癥狀;未及淺表淋巴結腫大,雙肺呼吸音低,未聞及干濕啰音,肝脾未捫及明顯腫大。門診胸部CT檢查提示“雙肺多發團塊及片狀高密度影,腫瘤性可疑,縱隔淋巴結增大”(圖1),擬“雙肺多發結節”收住入院。患者有“高血壓”疾病史,30年前因“附睪結核”行附睪剝離術,術后正規抗結核治療后治愈,有吸煙史40支×40年。實驗室檢查:血常規:WBC 2.8×109/L,N 81.9%,L 11.2%,HGB 119G/L,PLT 142×109/L;病毒抗體測定提示EBV-IgM陽性;血氣分析:pH 7.43,PaO270mmHg,PaCO240mmHg;血沉39mm/h,CRP 1.03mg/dl。風濕免疫、ANCA、腫瘤標志物、PPD、痰培養、血培養、痰找抗酸桿菌、支氣管鏡毛刷涂片找抗酸桿菌、脫落細胞等檢查均為陰性。支氣管鏡檢查提示“氣管支氣管炎癥改變”;肺功能檢查提示“輕度混合性通氣功能障礙,小氣道功能障礙,彌散功能輕度降低,氣道阻力正常”。胸部增強CT檢查(距離門診胸部CT檢查僅5d)提示“雙肺多發團塊及片狀高密度影,病灶有所進展,考慮惡性腫瘤,淋巴瘤可能,肺癌待排,建議穿刺活檢,縱隔淋巴結腫大,脾臟增大”。
完善相關檢查后對該患者先后進行2次肺穿刺活檢,第1次肺穿刺部位針對左下肺結節,病理穿刺結果提示“慢性肉芽腫性炎,抗酸染色(-),PAS(-),六胺銀(-)”。與家屬溝通后進行第2次肺穿刺,針對右上肺實變區域。穿刺過程中出現致命大咯血,經搶救后生命體征平穩。穿刺病理結果提示“肺實變,大量T淋巴細胞散在B細胞,并見浸潤血管壁,考慮淋巴細胞增生性病變”。免疫組化:CD20(+),CD3(+),CD4(-),CD43(+),CD56(+),CD68(+),CD79a(+),Granzyme B(-),Ki-67(+)40%,Perforin(-),TdT(-),TIA-1(+)。特殊染色:抗酸染色(-),PAS(-),六胺銀(-)。結合病理結果,診斷為淋巴瘤樣肉芽腫,病理分型為I型(圖2)。
根據病理分型,予強的松60mg/d口服治療,1個月后病灶吸收理想(圖3),第2個月減量至45mg/d,復查發現病灶大部分吸收,第3個月再減量至30mg/d,此劑量共維持2個月。病灶基本吸收,未復發(圖4)。病程中出現發熱1次,體溫最高39℃,胸部CT復查未出現新病灶,考慮因免疫功能減低致上呼吸道感染,經抗感染等對癥治療后好轉,復查血常規WBC 4.2×109/L,之后根據復查情況逐漸減少糖皮質激素用量,目前已隨訪10個月,維持劑量10、15mg隔日1次,胸部CT檢查提示病灶繼續吸收中,未出現新的病灶,患者精神狀態佳,輕度庫欣面容,繼續隨訪中。
討論淋巴瘤樣肉芽腫(LYG)從病理角度闡述是一種由EBV感染引起的非典型B細胞混合大量反應性T細胞和組織細胞所組成的結外血管中心性和血管破壞性淋巴組織增生性疾病。Liebow等在1972年首先報道了本病。本病好發于男性,病變進展快,可侵犯全身多種臟器,主要累及肺、皮膚、中樞神經系統等。影像學主要表現為雙肺多發結節、腫塊,多數位于下肺,邊緣模糊呈絨毛狀,直徑0.5~8cm,常合并壞死、空洞,可見支氣管征,肺門、縱隔淋巴結腫大少見,可見胸腔積液。本例患者以咳嗽、咳痰為主要臨床癥狀,胸部CT檢查表現為兩肺多發團塊狀、片狀、結節融合、滲出,且病情進展迅速。
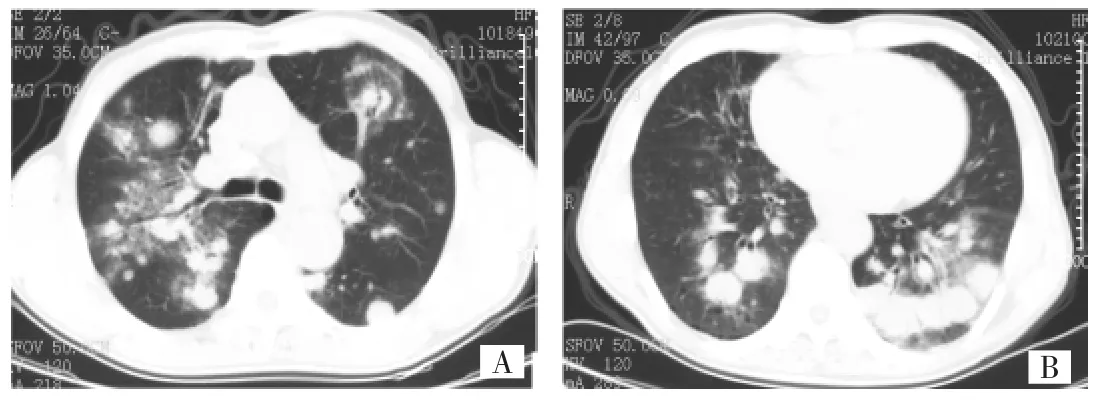
圖1 治療前胸部CT檢查所見(A:隆突平面可見雙肺多發團塊及片狀高密度影,病變形態出現多樣性;B:近心底層面雙肺基地節隔間多發團塊狀病變,并呈融合趨勢)

圖2 肺穿刺活檢結果(A:病變主要由混合性小淋巴細胞和少量不典型性大單核淋巴樣細胞構成,大細胞類似免疫母細胞,有明顯核仁,并見浸潤血管壁,HE染色,高倍;B:不典型性大單核淋巴細胞示CD20陽性,En Vision法,高倍;C:不典型性大單核淋巴細胞示CD79a陽性,En Vision法,高倍)
該病診斷困難,臨床癥狀和影像學表現與Wegener肉芽腫相似,肺部出現結節狀病灶時,臨床上易與結核、肉芽腫病、肺癌及炎性假瘤等疾病相混淆,病理上應與結核、非特異性肉芽腫病、肺外的外周T細胞淋巴瘤等鑒別,形態學、免疫表型結合臨床特征可明確診斷。本病預后差,10%~60%患者會進展為大B細胞淋巴瘤,據報道中位生存期為24個月[1]。

圖3 治療1個月后胸部CT檢查所見(A:隆突平面可見雙肺多發團塊及片狀高密度影吸收理想;B:近心底層面雙肺基底節隔間多發團塊病變明顯縮小)

圖4 治療3個月后胸部CT檢查所見(A:隆突平面可見雙肺多發團塊及片狀高密度影基本吸收;B:近心底層面雙肺基底節隔間多發團塊病變基本消失)
治療上,因本病罕見,尚未建立標準的治療方案,對于肺部病變較為局限的患者,多主張積極手術治療,術后可行全身系統治療,藥物治療主要根據組織學分級來選擇[2]。本例患者在診斷明確、征得患者及家屬同意簽字情況下,采用長療程、足劑量糖皮質激素治療,目前已隨訪10個月,糖皮質激素治療效果理想,病灶明顯吸收,且未見復發。Sebire等[3]于2003年首次報道了單用利妥昔單抗治療LYG有效的報道。據目前應用利妥昔單抗治療并報道的12例患者中成功9例,失敗3例[4]。可見利妥昔單抗治療不失為一個好的選擇及方向。目前,本例患者仍在隨訪中,糖皮質激素治療效果理想,但不能排除今后復發可能,必要時可以考慮利妥昔單抗治療,但費用昂貴,需征得患者及家屬同意。
[1]CulhaciN,LeviE,Sen S,et al.Pulmonary lymphomatoid granulomatosis evolving to large cell lymphoma in the skin[J]. Pathology and Oncology Research, 2002,8(4):280-282.
[2]Ishiura H,Morikawa M,Hamada M,et al. Lymphomatoid granulomatosis involving centralnervous system successfully treated with rituximab alone[J].Arch Neurol,2008,65(5):662-665.
[3]Sebire N J,Haselden S,Malone M,et al. Isolated EBVlymphoproliferative disease in a child with Wiskott-Aldrich syndrome manifesting as cutaneous lymphomatoid granulomatosis and responsive to anti-CD20 immunotherapy[J].J Clin Pathol, 2003,56(7):555-557.
[4]Castrale C,El Haggan W,Chapon F,et al.Lymphomatoid granulomatosis treated successfully with rituximab in a renal transplant patient[J].J Transplant,2011, 2011:865-957.
(本文編輯:嚴瑋雯)
《浙江醫學》“病例討論”欄目征稿
根據廣大讀者的建議,本刊自2010年第7期起開辟“病例討論”欄目,論文結構分為“病例摘要”和“討論”兩部分,以期通過對疑難、復雜或罕見病例的介紹和討論,交流臨床工作經驗,幫助廣大臨床醫師掌握科學的臨床思維方式,提高各專科和多學科的綜合分析判斷能力,進而提高醫療水平。現特向廣大臨床醫師征集相關病例,具體要求如下。
1病例選擇(1)疑難病例,特別是涉及多學科、多領域的疑難病例;(2)診斷明確,但病情危重和(或)治療棘手的病例;(3)臨床較罕見的病例。以上病例均需最終獲得明確診斷或成功治療,且臨床資料齊全,并能提供實驗室、影像學和(或)病理確診依據。
2寫作格式和要求(1)病歷摘要:分段敘述患者的簡要病史(包括主訴、現病史、既往史等)、入院后體檢情況、輔助檢查結果、入院后治療方案及病情變化等內容;(2)討論:分段記錄各級或各科或各院醫師對該病例的特點、診斷、鑒別診斷、進一步輔助檢查和治療方案等方面的分析,若為罕見病,則需介紹目前國內外關于該病診治方面的最新進展;(3)列出相關的國內外主要參考文獻;(4)全文字數在3000字左右。
3投稿注意事項 投稿時請務必在稿件末頁留下第一作者手機號碼和電子郵箱地址,同時附上單位證明(證明該病例所有資料屬實,無一稿兩投,無涉及保密等情況)。
本刊編輯部
2012-04-23)
315040 寧波大學附屬李惠利醫院呼吸內科(宋美君、吳宏成、林志輝),病理科(潘登)
宋美君,E-mail:smj800722@ yahoo.com.cn
