熒光衍生法測定中藥二氧化硫殘留量研究
2013-04-22 02:28:40彭月,李雪蓮,銀玲,陳鴻平,樊丹青,劉友平
中國中藥雜志 2013年2期
關鍵詞:中藥
彭月,李雪蓮,銀玲,陳鴻平,樊丹青,劉友平
[摘要] 目的:采用衍生化反應,建立熒光分光光度法測定中藥中二氧化硫的殘留量。方法:對影響熒光強度的因素進行研究,確定最佳衍生條件。在激發(fā)波長321 nm,發(fā)射波長384 nm處測定熒光強度。結果:熒光強度與Na2SO3對照品加入量在0.999 7~17.99 nmol線性關系良好,r=0.999 9,平均回收率102.3%,RSD 4.6%。結論:該法具有簡便易行,快速準確,靈敏度高等特點,可為中藥材二氧化硫殘留量的測定提供參考。
[關鍵詞] 熒光分光光度法;衍生;二氧化硫殘留量;中藥
硫磺是一種古老的用于中藥防蟲的藥物,不少中藥材產地加工時都會硫熏,利用硫磺加熱產生的二氧化硫達到漂白藥材及殺菌防霉防蟲的目的,由于硫磺熏蒸后中藥材的性狀及化學成分多有改變,且熏蒸后的中藥材中殘留的二氧化硫對人體有很大傷害[1],《中國藥典》2005年版取消了藥材的“硫磺熏蒸養(yǎng)護法”, 并在《中國藥典》2010年版附錄中增加了基于蒸餾法的二氧化硫殘留量測定法,由于該法要求專門的全玻璃蒸餾儀器,推廣很慢,其他測定中藥二氧化硫殘留量的方法尚有鹽酸副玫瑰苯胺法[2]、高效液相色譜法[3]、氣相色譜法[4]和離子色譜法[5]等,但鹽酸副玫瑰苯胺法操作過程中需使用有毒的四氯汞鈉溶液,幾種色譜法需要相應的設備及填充柱等,檢測成本較高。
本研究根據亞硫酸鈉-鄰苯二甲醛-銨鹽在中性或弱酸性條件下反應生成具有強烈熒光的化合物1-磺酸基-異吲哚,首次建立了熒光衍生法用于中藥材二氧化硫殘留量測定的方法,具有靈敏度高,選擇性好,分析速度較快等優(yōu)點,以期為中藥材二氧化硫殘留量的測定提供一種新的參考方法。
1 材料
RF-5301(PC)S熒光分光光度計(日本島津公司);電子分析天平BP211D(1/10 萬,德國Sartorius公司),電子分析天平BP121S(1/1 萬,德國Sartorius公司);pHS-3C型酸度計(上海雷磁儀器廠);WHY-2水浴恒溫振蕩器(江蘇金壇市金城國勝實驗儀器廠);恒溫水浴鍋W201B(北京國華醫(yī)療器械廠)。
亞硫酸鈉對照品(美國Fisher公司,批號120224,純度99.5%);其他試劑均為分析純,實驗用水為重蒸餾水;中藥材樣品購于荷花池中藥材市場,均經成都中醫(yī)藥大學盧先明教授鑒定。
2 方法
2.1 試劑配制
亞硫酸鈉對照品儲備液:精密稱取亞硫酸鈉對照品適量,加水溶解制成1 mmol·L-1亞硫酸鈉對照品儲備液,避光冷藏,使用時用水稀釋至所需濃度。
鄰苯二甲醛溶液:準確稱取0.134 1 g鄰苯二甲醛,無水乙醇溶解后定容至100 mL棕色量瓶中作為儲備液,避光冷藏,使用時移取10.0 mL定容至100 mL配成1 mmol·L-1的鄰苯二甲醛工作液。
乙酸銨溶液:準確稱取7.708 0 g乙酸銨(AR),用水溶解后定容至1 000 mL棕色量瓶中作為儲備液,避光冷藏,使用時移取5.0 mL定容至100 mL配成5 mmol·L-1乙酸銨工作液。
磷酸氫二鈉-磷酸二氫鉀緩沖溶液:將濃度為0.3 mol·L-1的磷酸氫二鈉和磷酸二氫鉀溶液按照不同體積比混合,配成一系列不同pH的緩沖溶液。
2.2 熒光衍生法測定
準確移取2.0 mL 磷酸氫二鈉-磷酸二氫鉀緩沖溶液(pH 6.44),適量樣品溶液于10 mL量瓶中,依次加入1 mmol·L-1鄰苯二甲醛溶液2.5 mL及5 mmol·L-1乙酸銨溶液1.5 mL,混勻,用水定容,于50 ℃水浴中恒溫5 min,取出,立即放入冰水中冷卻,終止反應,靜置40 min,以試劑空白為參比,于1 cm的石英比色皿中測定激發(fā)波長為321 nm,發(fā)射波長為384 nm時樣品的相對熒光強度。
3 結果
3.1 激發(fā)光譜和發(fā)射光譜
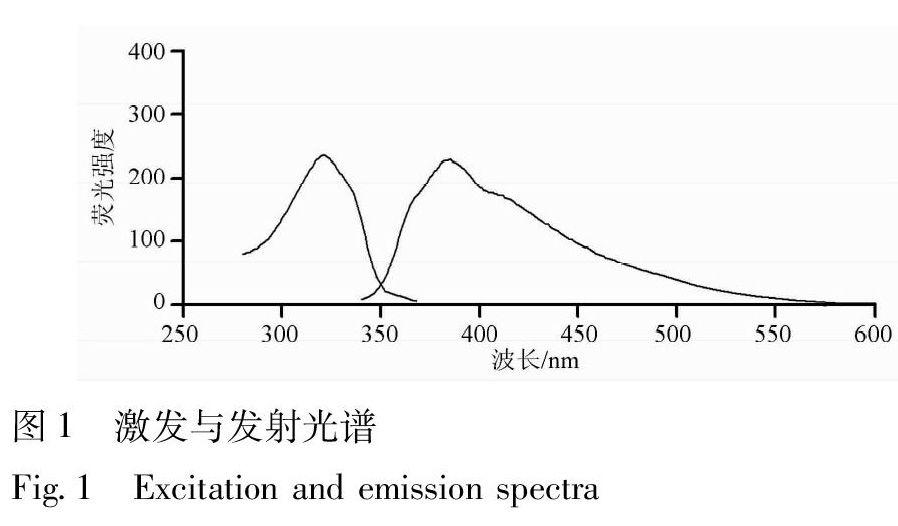
按實驗方法配制溶液,然后在熒光分光光度計上分別掃描其激發(fā)光譜和發(fā)射光譜,見圖1,可知體系的最佳激發(fā)波長為321 nm,發(fā)射波長為384 nm。
3.2 試驗條件的選擇
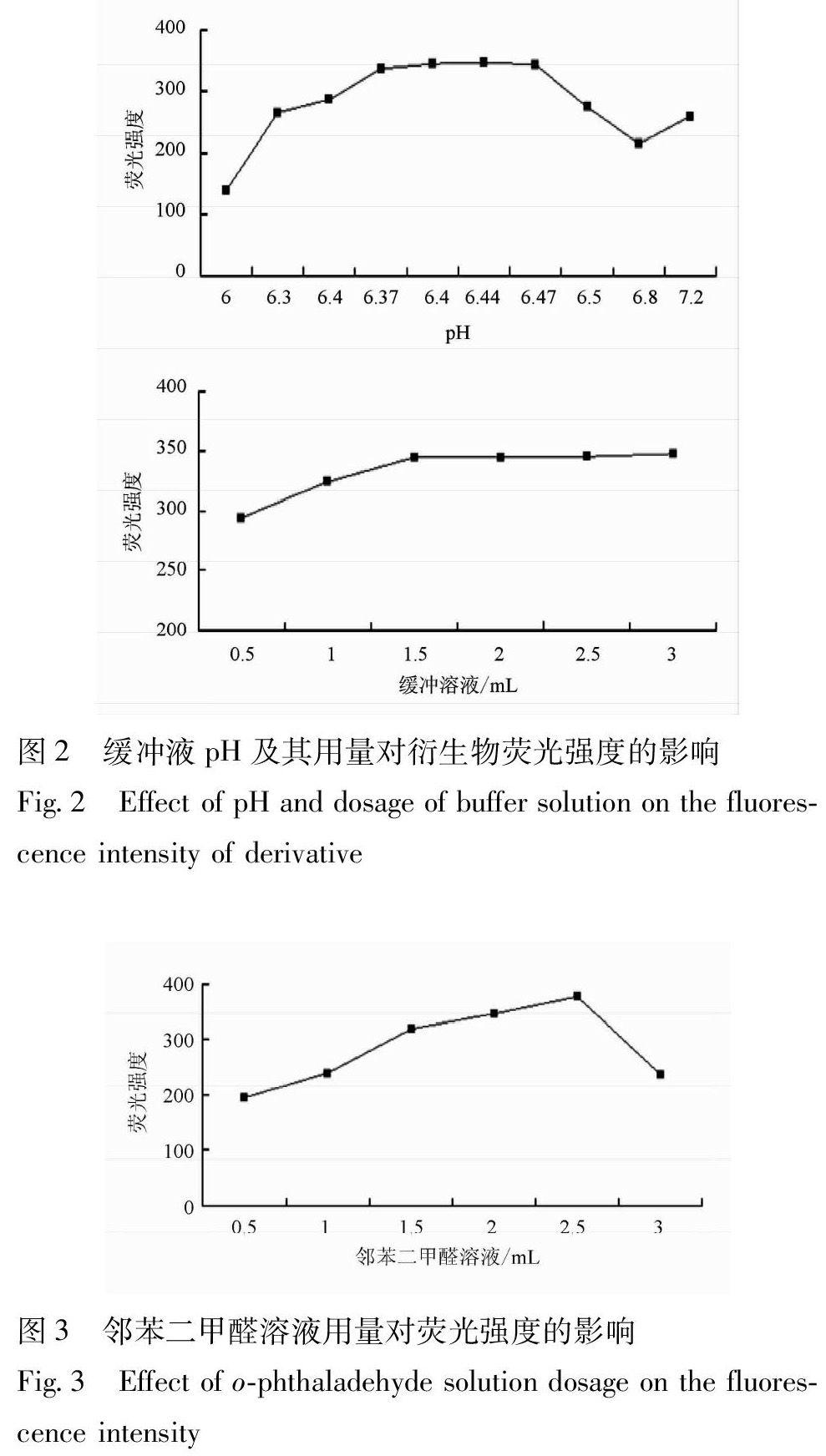
3.2.1 緩沖液pH及其用量的影響 試驗考察了不同pH 磷酸氫二鈉-磷酸二氫鉀緩沖溶液對體系熒光強度的影響,見圖2,發(fā)現(xiàn)pH在6.37 ~ 6.47,衍生物的相對熒光強度大且穩(wěn)定。當pH為6.44,產物的相對熒光強度最大,故選擇pH 6.44的磷酸氫二鈉-磷酸二氫鉀緩沖溶液作為測定的酸度條件,其加入量2.0 mL較為適宜。
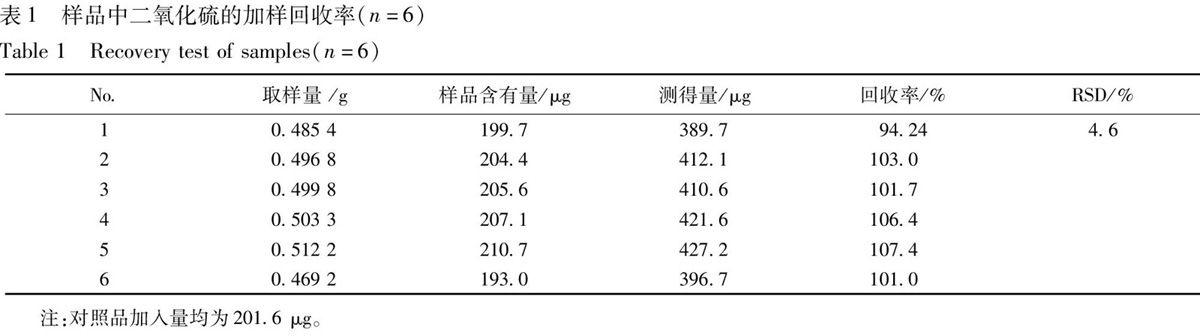
3.2.2 鄰苯二甲醛用量的影響 取適量的亞硫酸鈉分別加入不同體積的1 mmol·L-1鄰苯二甲醛溶液,經衍生后測定產物的相對熒光強度,見圖3,表明隨著鄰苯二甲醛溶液用量的增大,產物的熒光強度先增大后降低,當其用量為2.5 mL時,熒光強度最大,繼續(xù)增加用量產物熒光強度反而降低,故本實驗選擇加入1 mmol·L-1鄰苯二甲醛溶液2.5 mL。
3.2.3 乙酸銨濃度及用量的影響 試驗考察了不同濃度乙酸銨對衍生物熒光強度的影響,見圖4,隨著乙酸銨濃度的增大,衍生物的熒光強度先增大后降低,當乙酸銨濃度為5 mmol·L-1時,熒光強度達到最大,故本實驗選擇5 mmol·L-1乙酸銨溶液,其加入量1.5 mL較為適宜。
3.2.4 反應溫度和反應時間的影響 按實驗方法,分別在不同溫度和反應時間下進行衍生反應,然后測定產物的相對熒光強度,見圖5,表明50 ℃水浴中反應5 min最佳,然后立即冰水浴冷卻,終止反應。
3.2.5 靜置時間的影響 按實驗方法對對照品試樣衍生化處理后,靜置一定時間后測定其相對熒光強度,見圖6,隨靜置時間延長產物相對熒光強度有所降低,當靜置在30~50 min衍生物相對熒光強度趨于穩(wěn)定,故本實驗選擇反應完成后靜置40 min測定產物的相對熒光強度。
3.3 供試品溶液的制備
取中藥細粉約1 g,精密稱定,精密加入0.5%氫氧化鈉溶液25 mL,搖勻,振蕩提取(200 r·min-1,40 ℃) 30 min,濾過,取1.0 mL稀釋至25 mL,即得。
3.4 方法學驗證
3.4.1 線性關系試驗 分別精密吸取對照品儲備液0.1,0.3,0.7,1.2,1.5,1.8 mL用水稀釋至100 mL,然后再分別移取上述不同濃度的稀釋液1.0 mL,按實驗方法測定產物的相對熒光強度。以熒光強度為縱坐標,亞硫酸鈉對照品的加入量(nmol)為橫坐標,繪制標準曲線,得回歸方程Y=46.247 X-15.007, r2=0.999 8,表明亞硫酸鈉加入量在0.999 7~ 17.99 nmol線性關系良好。
3.4.2 精密度試驗 取同一批山藥樣品,按3.3項下方法,制備供試品溶液,平行測定6次,記錄熒光強度,計算RSD 1.6%,表明儀器精密度良好。
3.4.3 穩(wěn)定性試驗 取已制備好的同一份樣品溶液,測定前避光冷藏,按上述實驗方法分別在0,0.5,1,1.5,2,2.5 h測定,記錄熒光強度,結果2 h內樣品熒光強度的RSD 2.8%,表明樣品溶液在避光冷藏的條件下2 h內穩(wěn)定。
3.4.4 重復性試驗 取同批次山藥樣品,按供試品溶液制備方法平行制備6份供試品溶液,按上述實驗方法測定其熒光強度,結果樣品中二氧化硫質量分數平均為 411.4 μg·g-1,RSD 1.9%,表明該方法重復性良好。
3.4.5 加樣回收試驗 取已知二氧化硫含量的同批次山藥樣品6份,約0.5 g,精密稱定,精密加入亞硫酸鈉對照品溶液適量,按供試品溶液的制備與測定同法操作,測定熒光強度,計算二氧化硫含量與回收率,得平均回收率為102.3%,RSD 4.6%,見表1。
3.4.6 樣品測定 按照建立的方法測定所購買部分中藥材的二氧化硫含量,見表2。
4 討論
基于亞硫酸鈉-鄰苯二甲醛-銨鹽在中性或弱酸性條件下反應生成具有強烈熒光的化合物1-磺酸基-異吲哚的原理,本研究建立了一種測定中藥中二氧化硫含量的新方法。較之《中國藥典》2010年版附錄規(guī)定的標準方法需特制的全玻璃蒸餾裝置,測定過程中人為、偶然誤差較大,該熒光衍生法操作過程可控,人為誤差小。本方法具有簡便、快速、靈敏度和準確度高等特點,對常見離子進行熒光光譜的干擾實驗結果表明,當亞硫酸鈉的加入量為12.00 nmol,相對誤差在5%內時,100倍的Na+,K+,NH4+,Cl-,HCO3-,NO2-,SCN-,H2PO4-,HPO42-,S2-,SO42-,CO32-等的存在對測定無干擾。利用該法對山藥、葛根、枸杞和麥冬的測定與2010年版《中國藥典》方法結果較一致,對于方法的測定結果可靠性的驗證有待進一步研究。
中藥硫熏過程中,單質硫經加熱氧化成二氧化硫,與中藥里的無機元素生成亞硫酸鹽,以游離態(tài)、可逆結合態(tài)和不可逆結合態(tài)3種形式存在。本研究采用堿性溶液提取樣品,可將其中可逆結合態(tài)的亞硫酸鹽釋放出來,并對振蕩提取法提取過程中的某些影響因素進行研究,結果確定的提取時間比標準方法大大縮短,利用此方法對山藥、葛根、枸杞和麥冬進行一次提取,與2010年版《中國藥典》規(guī)定方法比較各藥提取率均在90%以上,表明振蕩提取法可作為一種樣品前處理方法用于中藥二氧化硫殘留量的測定。該法采用儀器提取的方式,自動化程度較高,適用于大批量樣品的提取。
本研究應用建立的熒光衍生法對部分中藥材中的二氧化硫含量進行測定,與中藥材中二氧化硫殘留量檢測限度征求意見稿規(guī)定的限量進行比較,購買的藥材中二氧化硫殘留量均不符合要求,提示目前中藥硫熏的現(xiàn)象仍然存在,對中藥材硫熏的替代加工技術有待深入研究。
[參考文獻]
[1] 劉東奇,陳華成,楊雪麗. 二氧化硫對機體各組織器官毒性作用的研究進展[J]. 獸牧獸醫(yī)雜志,2008,27(1):37.
[2] 中國國家標準管理委員會. 食品衛(wèi)生檢驗方法理化部分(一)[S]. GB/T5009.34-2003: 267-273.
[3] Stephen Wai Cheung Chung, Benny T P Chan, Andy C M Chan. Determination of free and reversibly-bound sulfite in selected foods by high-performance liquid chromatography with fluorometric detection[J]. J AOAC Int,2008,91:98.
[4] 孫艷平,李濤. 頂空氣相色譜火焰光度檢測器檢測山藥中的二氧化硫[J]. 山西醫(yī)學雜志, 2010,39(8):1024.
[5] 王欣美,夏晶,王柯,等. 堿性溶液提取-離子色譜法測定中藥中二氧化硫殘留量[J]. 中國中藥雜志,2011,36(19):2656.
Determination of sulfur dioxide in traditional Chinese
medicine by derivative fluorometry
PENG Yue, LI Xue-lian, YIN Ling, CHEN Hong-ping, FAN Dan-qing, LIU You-ping*
(Laboratory Constructed by PRC and Sichuan Province in Systematically Research and Development
Utilization of Traditional Chinese Medicine Resources, Department of Pharmacy, Chengdu
University of Traditional Chinese Medicine, Chengdu 611137, China)
[Abstract] Objective: To establish a derivative fluorometry method for the determination of sulfur dioxide residues in traditional Chinese medicine. Method: The optimal derivation condition was established. The fluorescence intensity was detected at excitation wavelength of 321 nm, and emission wavelength of 384 nm. Result: A linear relationship was obtained between the fluorescence intensity and the addition of reference substance in the range of 0.999 7-17.99 nmol with a correlation coeffient of 0.999 9, and the average recovery was 102.3% with RSD 4.6%. Conclusion: This method is simple and sensitive with quick and correct result. It can provide a reference for the determination of sulfur dioxide residues in traditional Chinese medicine.
[Key words] fluorescence spectrophotometry; derivative; sulfur dioxide residues; traditional Chinese medicine
doi:10.4268/cjcmm20130214
[責任編輯 孔晶晶]
猜你喜歡
中老年保健(2021年5期)2021-12-02 15:48:21
中老年保健(2021年4期)2021-12-01 11:19:40
中老年保健(2021年4期)2021-08-22 07:08:32
中國現(xiàn)代中藥(2020年10期)2020-12-16 08:53:18
金橋(2020年7期)2020-08-13 03:07:00
基層中醫(yī)藥(2020年12期)2020-07-22 06:34:38
中國現(xiàn)代中藥(2020年4期)2020-06-10 09:56:34
基層中醫(yī)藥(2018年6期)2018-08-29 01:20:20
長春中醫(yī)藥大學學報(2017年1期)2017-04-16 05:56:49
肝博士(2015年2期)2015-02-27 10:49:49
