二十多種遺傳性骨病的快速鑒別診斷
2013-07-08 02:17:10郭奕斌
分子診斷與治療雜志 2013年2期
郭奕斌
?述 評?
二十多種遺傳性骨病的快速鑒別診斷
郭奕斌
遺傳性骨病種類繁多,臨床上相對常見的也有二、三十種。這些骨病癥狀、體征相似,難以鑒別,分子診斷時間長、費用高,若無法進行有針對性的基因檢測或蛋白/酶功能鑒定,勢必造成時間和經費的損失。本文根據十多年來對二十多種遺傳性骨病、代謝病進行分子診斷和早期防治工作所積累的一些診防經驗,對各病的快速鑒別要領進行了比較全面的分析總結,以期對臨床診、防、治工作提供一些有益的啟示。
遺傳性骨病;鑒別診斷;快速
遺傳性骨病種類繁多,根據2010年修訂的國際遺傳性骨病的最新分類標準,一共分為40組456種[1]。臨床上相對常見的有:軟骨發育不全(ACH)、軟骨發育低下(HCH)、致死性侏儒癥(TD)、假性軟骨發育不全(PSACH)、多發性骨骺發育不良(MED)[2~4]、先天性脊柱骨骺發育不良(SEDC)、遲發性脊椎骨骺發育不良(SEDT)、X連鎖低血磷性佝僂病(XLH)、軟骨生成不全(ACG)、短肋多指(趾)綜合征(SRPS)、黏多糖貯積癥(MPS,有7大型17種亞型)[5]、成骨不全(OI,有I~VIII 8型)[6~7]、窒息性胸廓發育不良(ATD,有5型)[8~9]等20多種。在這些骨病中,除TD、OI-II、ACG、SRPS胎死腹中或出生不久即夭折外,其他骨病都有個頭矮小、骨骼畸形、癥狀進行性加重、致殘甚至致死等癥。它們彼此之間如MPS I和MPS II,MPS I和MPS VI,ACH和HCH,ACH和PSACH,PSACH和MED,TD和OI,SEDT和SEDC等都有諸多相似之處,僅憑臨床癥狀通常難于鑒別,往往需分子診斷(包括基因診斷和(或)蛋白/酶學功能鑒定)方能確診,而只有確診才能為后續的產前/植入前診斷和治療創造前提條件。而分子診斷一來過程復雜、時間長(短則數星期,長則數個月甚至大半年),二來試劑、檢測費用高。如能掌握各病的特征和要領,進行快速的初步鑒別,然后確定最可能的病種,再進行有針對性的DNA和(或)RNA和(或)蛋白檢測,不但可以省下相當可觀的試劑費和檢測費,更重要的是可以省下用于患兒早期治療和孕婦早期產前診斷的時間。根據十多年來對二、三十種遺傳性骨病、代謝病進行分子診斷和早期防治工作所積累的一些診防經驗,現把各病的快速鑒別要領歸納總結如下。圖1列出了十種遺傳性骨病的主要臨床特征。
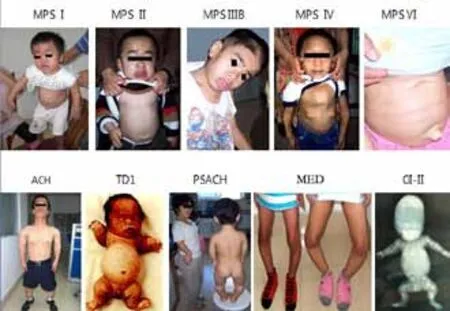
圖1 十種遺傳性骨病的主要臨床特征Figure 1 The main clinical feature of ten kinds of genetic skeletal disorder
1 黏多糖貯積癥各型之間的快速鑒別
1.1 MPS I型(Hurler綜合征)和MPS II型(Hunter綜合征)的鑒別
兩者癥狀和體征(包括個頭矮小、智力中至重度受損、骨骼畸形、特殊面容、肝脾腫大、臍疝、蒙古斑、爪形手等)都很相似,尿糖胺聚糖(GAGs)檢測結果也很一致,都是強陽性(++或+++),所以僅從外觀和尿檢結果很難鑒別。但下列三點對快速鑒別很有幫助:①首先確認患者是男性還是女性?若是男性,再確認是否有家族史?若有,再確認所有患者是否都是男性?是否都是母系的男性親屬發病?若答案是肯定的話,那II型的可能性極大,直接查IDS基因即可。②如果沒有家族史,但患兒是男性,那首先看下患兒的角膜是否混濁?如果角膜明亮,則II型可能性大,應先查IDS基因。③如果患兒是女性,則II型的可能性極小,幾乎可以排除是II型,應先查I型的IDUA基因。
1.2 MPS I型和MPS III型(Sanf i lippo綜合征)的鑒別
如果患兒都是女性或都是男性,又沒家族史,尿GAGs檢測又都呈強陽性,此時主要看智商和體征。III型患兒通常智力嚴重受損,受損程度可達(++++),而其他體征則基本正常,個頭不但不矮甚至超過同齡小孩,骨骼也沒有畸形,角膜也不混濁[10];而I型除了智力嚴重低下(+++)外,還有個頭矮小(比同齡小孩矮半個頭至一個頭),角膜混濁,而且骨骼畸形,爪形手,橈尺骨僵硬,走路呈鴨步;背部臀部蒙古斑多甚至成片狀等。
1.3 MPS I、II、III型與MPS IV型(Morquio綜合征)的鑒別
MPS IV型的智力正常或基本正常,尿檢多半是弱陽性(+/-)或陽性(+)且有KS帶,癥狀主要表現在骨骼畸形嚴重,可有雞胸、肋骨外翻,腰椎后凸、駝背,膝內翻呈X形腿等,這一點可以和典型的I型、II型、III型相鑒別。
1.4 MPS I型和MPS VI型(Maroteaux-Lamy綜合征)的鑒別
兩者癥狀/體征、系譜分析和尿GAGs檢測結果都很相似,所以僅從外觀、遺傳方式和尿檢結果很難鑒別。但有一點對于快速鑒別很有幫助,就是確認患兒的智力水平。如果智力受損嚴重、智商很低,則可以排除VI型,應先查I型的IDUA基因;反之,如果智力正常或基本正常,則應先查VI型的ARSB基因。
1.5 MPS II型和MPS III型的鑒別
如果患兒都是男性,且都沒家族史,角膜也不混濁,尿檢也都呈強陽性,此時主要看其智商和體征。兩者的鑒別基本同1.2。
1.6 MPS II型與MPS VI型的鑒別
兩者都有頭大、頸短、特殊面容、臍疝、蒙古斑多、橈尺骨僵硬,尿檢都呈強陽性。若都是男性患兒,則系譜分析是鑒別診斷第一要素。若無家族史,則測智商、驗角膜是鑒別兩者的關鍵。II型智力受損嚴重(+++),但角膜明亮;而VI型智力正常或基本正常,但角膜與I型類似,混濁明顯。
1.7 MPS IV型與MPS VI型的鑒別
兩者智力都正常或基本正常。但VI型有特殊面容(頭大、頸短、濃眉、額凸、鼻梁凹、角膜混濁、鼻翼大、嘴唇厚、牙齒稀疏),腹大,臍疝明顯,個矮,體毛多,蒙古斑多,行走困難,尿檢呈強陽性(++或+++);而IV型則主要表現在雞胸、肋骨外翻、膝內翻、腰椎后凸,背部隆起,角膜多不混濁,尿檢多呈弱陽性(+/-)或至多陽性(+)。
2 軟骨發育不全與其它骨軟骨病的快速鑒別
2.1 ACH與HCH的鑒別
都是FGFR3基因突變所致,都是短肢型侏儒,軀干發育都近似正常,上部量都大于下部量。但ACH出生時即可見特殊面容,且因胎兒頭大,多伴有剖宮產史。多數ACH有三叉狀或車輪狀手,站立時常見“O”形腿,臀部后凸。本病可通過超聲和(或)基因分析進行產前診斷。而HCH患兒雖身材也矮小,但較ACH為輕,頭顱和面部相稱,無明顯特殊面容,部分病例伴膝內翻或肘外展受限。2.2 ACH與TD胎兒的鑒別
兩者也都是FGFR3基因突變所致。但對于ACH胎兒來說,B超通常都是在孕晚期(30~40周)才超出有異常,且僅發現長骨粗短,而TD患胎通常在孕中期(21~27周)即發現有異常,且除發現四肢長骨明顯短小、呈蛙式體態外,還發現胸廓明顯狹窄。
2.3 ACH與PSACH的鑒別
兩者都有個頭矮小,O型腿,智力和頭面部發育正常等相似癥狀,身體比例也很相似。但PSACH患兒出生時發育正常,一般在2歲后四肢近端才開始短縮,到成人時身高為106~130 cm。患兒四肢關節雖明顯增大,手指粗短,但無典型三叉指形。本病由于胎兒期及出生時外觀正常,故不能通過超聲檢查進行產前診斷,僅能通過相關基因的突變檢測進行產前診斷。而ACH出生時即表現為頭顱大,四肢短,三叉手。另外,ACH可通過超聲進行產前診斷。
2.4 ACH與OI的鑒別
ACH患兒一般無骨折,骨皮質也不變薄,甚至增厚;而成骨不全患兒易出現骨折及骨皮質變薄,且有特征性的藍色鞏膜癥狀,此可能為OI-I型或OI-IV型。在孕期,如果B超超出有長骨骨折痕跡,則患OI-II的可能性極大。
2.5 ACH與MPS IVA型的鑒別
兩者的體征有諸多相似之處如個頭矮小、智力正常,且尿GAGs定性檢查常為弱陽性(+/-)或陽性(+),此時主要看胸骨、肋骨、腰椎的病變情況。如果骨骼畸形嚴重特別是有雞胸、肋骨外翻、膝內翻呈X形腿、腰椎后凸、背部隆起,則為MPS IVA型。而ACH多呈O形腿、三叉手、特殊面容、臀部后凸,ACH可在孕期通過超聲或基因檢測進行產前診斷。而MPS IVA型,孕期無法通過B超檢查,僅能通過GALNS基因的產前基因診斷。
2.6 ACH與XLH的鑒別
兩者都有O形腿、佝僂病癥。但ACH為常染色體顯性遺傳,男女患病率相同,且病情相似,可以通過B超進行產前診斷。而XLH是X連鎖顯性遺傳,女性患兒多于男性患兒,但女性患兒較輕,男性患兒較重,且XLH無法通過B超進行產前診斷。
3 其他骨軟骨病之間的快速鑒別
3.1 PSACH與MED的鑒別
兩者相似度很高,且主要都是由COMP基因突變所致,只是突變部位不同,表現程度不同而已。輕型PSACH與MED存在表型的重疊,但一般較MED來得嚴重。PSACH個頭很矮,一般不超過130 cm,而MED可達145~170 cm,且屬于勻稱性矮小。
3.2 TD與OI-II的鑒別
兩者都是導致胎兒死亡的原因。TD常合并智力低下、心臟畸形和腎盂發育缺陷等,在孕期除了四肢長骨明顯短小外,還有胸廓明顯狹窄,本病系FGFR3基因異常所致。而OI-II除了四肢短、胸廓窄外,還有明顯骨折痕跡,是COL1A1基因或COL1A2基因突變所致。
3.3 TD1與TD2的鑒別
TD1患兒的突出表現是股骨彎曲畸形,呈電話聽筒狀;而TD2無股骨畸形,但存在顱骨畸形,患兒表現為頭大,前額突出,“三葉草”樣頭顱,鼻嵴低平,四肢短小,胸部發育不全。
3.4 TD與ATD的鑒別
ATD表現為四肢粗短,胸廓小而狹長,生后即有呼吸困難;肋骨短,水平排列,手足短而寬,多指(趾),鎖骨高位,雙肩峰端寬闊,張開如自行車把式,為AR遺傳,父母常為攜帶者。而TD雖也四肢粗短,胸廓狹窄,但頭大,四肢呈蛙式體態,肌張力低下,原始反射、腱反射消失,為AD遺傳,常為散發病例,是FGFR3基因新生突變所致。
3.5 TD與ACG的鑒別
兩者臨床癥狀體征十分相近,以頭大、短肢畸形、皮膚堆積成皺、肝脾腫大、出生后短時間內即死亡為其共同特點。很容易與其他短肢侏儒如ACH、ATD等混淆。二者主要區別是:(1)軀干長短不同:ACG短,只有24 cm左右;而TD正常,可達35 cm。(2)TD可在胎內由B超發現腦積水及三葉草狀頭顱,偶爾合并腦疝,而ACG無此征;(3)影像學區別:①ACG椎體扁平呈H形,骨盆骨化缺乏或嚴重延遲;而TD椎體呈“U” 字形,骨盆骨化存在。②ACG長管狀骨短,以肱骨、髂骨明顯,但不彎曲。而TD長管狀骨短且彎曲,以肢骨顯著,呈“電話聽筒”樣。
3.6 SEDT與SEDC的鑒別
SEDT主要表現為非勻稱性矮小,椎體畸形,早發的大關節炎,尤其在髖部,一般3~10歲才開始出現腰背部疼痛,生長遲緩,10~14歲最明顯,指距大于身高,上部量小于下部量。而SEDC主要表現為:脊椎骨骨骺發育不良,脊柱后凸,腰椎前凸,桶狀胸,胸骨隆凸。近視,視網膜剝離,腭裂,肌肉軟弱,易疲勞,腹肌發育不良。本病發病年齡早。
3.7 SEDT與MED的鑒別
兩者病變均累及脊柱和長管狀骨,但前者脊柱受累較重,長管狀骨受累輕,多為XR遺傳,家族中常有多代、多名男性患者,易于鑒別。而后者相反,脊柱受累輕,長管狀骨受累重,為AD遺傳,男女均可患病。
3.8 SEDT與MPS IVA的鑒別
MPS IVA型與SEDT都表現為短軀干型矮小,尿GAGs定性檢測均呈弱陽性或陽性,因此兩者易混淆。但MPS IVA型主要表現為雞胸,肋骨外翻,腰背部無痛性后凸,X形腿,多無家族史。而SEDT患者面容基本正常,以桶狀胸多見,腰椎無明顯后凸,背不痛,但多有家族史且患者多為男性。
3.9 先天性OI與遲發性OI的鑒別
前者屬嚴重型,出生時就有多發性骨折,產程中或子宮內的輕微外傷就可引起骨折。肢體短,有畸形并有摩擦音,顱骨如膜狀,此型患兒常因顱內出血而成死胎。而后者出生時表現正常,重癥者在嬰兒期可發生骨折,輕型發生骨折較晚,最輕者只有鞏膜發藍而不發生骨折。
3.10 SRPS I與SRPS II的鑒別
SRPS I型主要表現為身材矮小,水腫外觀,肋骨短、呈平行型,胸廓狹窄,四肢明顯縮短像潛水員用的泳蹼,心臟畸形,軸后多指(趾)。而II型除此之外,還有明顯的唇裂、腭裂。
3.11 XLH與營養性佝僂病的鑒別
XLH多在1歲左右起病,首發癥狀多為骨骼畸形,尤以下肢“O”型腿或“X”型腿多見。有家族史或雖無家族史,但家族中多有低血磷者。本病并非維生素D缺乏,而是由于近端腎小管對磷重吸收發生缺陷,使尿磷排泄增加、血磷下降所致。血鈣水平大致正常,一般劑量維生素D制劑不能提高血鱗[11]。而營養性佝僂病(也稱維生素D缺乏性佝僂病)是由于缺乏維生素D所致,故補充維生素D可以取得明顯療效。
3.12 ACG-I型與ACG-II型的鑒別
均為致死性新生兒軟骨發育不良,軟骨嚴重營養障礙,腰椎骨化不足,骶骨、恥骨和坐骨不骨化,四肢短型侏儒。但I 型癥狀更嚴重,身材更矮小,平均身長僅20~30 cm,且有肋骨薄,多發性骨折。而II型較輕,通常表現為扁平臉,巨大的顱蓋骨和大的前囟、后囟,腹部膨隆有腹水。
[1] Warman M L, Cormier-Daire V, Hall C, et al. Nosology and classif i cation of genetic skeletal disorders: 2010 revision[J]. Am J Med Genet Part A, 2011, 155 A(5): 943-968.
[2] 郭奕斌, 郭春苗, 王晶晶, 等. 遺傳性侏儒及其鑒別診斷[J].中國優生與遺傳雜志, 2007, 15(11): 117, 131.
[3] 張彩鳳, 郭奕斌. 軟骨發育不全與假性軟骨發育不全的分子遺傳學進展[J]. 熱帶醫學雜志, 2007, 7(93): 187-191.
[4] 王晶晶, 郭奕斌. 假性軟骨發育不全、多發性骨骺發育不良的分子遺傳學研究進展[J]. 遺傳, 2008, 30(5): 537-542.
[5] 郭奕斌. 黏多糖病的診斷及防治策略[J]. 中華臨床醫師雜志: 電子版, 2009, 3(10): 1620-1627.
[6] 艾陽, 唐佳, 方群, 等. 一例成骨不全II型高危胎兒的產前基因診斷[J]. 中華臨床醫師雜志: 電子版, 2011, 5(22): 6662-6666.
[7] 郭奕斌, 艾陽, 蔣瑋瑩. 一罕見成骨不全IV型的基因診斷[J]. 分子診斷與治療雜志, 2013, 5(1): 12-14.
[8] Marla J.F. O'Neil. Asphyxiating thoracic dystrophy 1[DB/ OL].(2011-2-12)[2012-9-20] http://omim.org/entry/208500.
[9] Keppler-Noreuil K M, Adam M P, Welch J, et al. Clinical insights gained from eight new cases and review of reported cases with Jeune syndrome (asphyxiating thoracic dystrophy) [J]. Am J Med Genet Part A, 2011, 155A(5): 1021-1032.
[10] Tang J, Pan J, Guo Y, et al. Mucopolysaccharidosis type IIIB mutations in Chinese patients: identification of two novel NAGLU mutations and analysis of two cases involving prenatal diagnosis[J]. Clin Chim Acta, 2013, 419: 33-38.
[11] 唐佳, 潘敬新, 蔣瑋瑩, 等. 一例抗維生素D佝僂病的基因診斷和新突變的致病性鑒定[J]. 中華臨床醫師雜志: 電子版, 2012, 6(5): 1226-1230.
Fast differential diagnosis of over twenty kinds of genetic skeletal disorders
GUO Yibin
(Department of Medical Genetics, Sun Yat-sen Medical College, Sun Yat-sen University, Guangdong, Guangzhou 510080, China)
The species of genetic skeletal disorders were very wide, and the relatively common species have twenty to thirty in the clinical. These skeletal disorders have similar signs and symptoms, diff i cult to identify, and molecular diagnosis would be long and costly. If targeted gene detection or protein/enzyme function identif i cation are incapable to be poformed, which would be cause the loss of time and money. According to the experience of molecular diagnosis and treatment of twenty kinds of genetic skeletal disorders and metabolic disease in the past decade, we make a relatively comprehensive analysis to rapid identif i cation for each disease in this paper, which was helpful for providing some benef i cial enlightenment to clinical diagnosis, prevention and treatment.
Genetic skeletal disorders; Differential diagnosis; Fast
國家自然科學基金(No.30772069);閩粵橫向課題基金(No.7101025)
中山大學中山醫學院醫學遺傳學教研室,廣東,廣州 510080
郭奕斌,E-mail: guoyibin@mail.sysu.edu.cn和zpgyp@yahoo.com.cn
