當歸調經膠囊的薄層鑒別與阿魏酸、芍藥苷的測定
2013-08-28 14:33:40謝仲德方應權李文烈夏天水
中成藥 2013年2期
謝仲德, 方應權, 李文烈, 夏天水
(1.重慶三峽醫藥高等專科學校藥學系,重慶萬州 404020;2.石家莊以嶺藥業股份有限公司,河北石家莊 050035;3.成都倍特藥業有限公司,四川成都 610041)
當歸調經膠囊是由當歸、熟地黃、川芎、黨參、白芍、甘草和黃芪組成的復方制劑。具有補血助氣,調經功效,用于貧血衰弱,病后、產后血虛以及月經不調,痛經[1]。是在原當歸調經顆粒[已收載于《部頒中藥成方制劑》第10冊 (WS3-B-1925-95)]的基礎上進行工藝改良而制備的新制劑,具有療效好、服用劑量小的特點。采用薄層色譜法對制劑中的當歸、川芎、白芍進行了定性鑒別研究,采用高效液相色譜法建立阿魏酸、芍藥苷的定量測定方法。
1 儀器與試藥
紫外分光光度計 (島津UV-3150)、恒溫水浴鍋 (余姚精益溫度儀表廠)、硅膠G(青島海洋化工有限公司)、高效液相色譜法儀 (北京創新通恒科技有限公司高效液相色譜儀:D3000高壓輸液泵,UV2000紫外分光檢測器)、電子天平 (BS-224S型,北京賽多利斯儀器系統有限公司)。當歸對照藥材 (批號9271-9905)、川芎對照藥材 (批號0919-200004)、阿魏酸對照品 (批號0773-9910)、芍藥苷對照品 (批號0736-200014)均購自中國藥品生物制品檢定所。化學試劑為分析純。當歸調經膠囊 (批號110201、110202、110203,自制)。
2 薄層鑒別
2.1 當歸、川芎的薄層色譜鑒別[2]取當歸調經膠囊樣品10粒,研細,取粉末2 g至燒杯中,加水10 mL溶解,加乙醇40 mL,邊加邊攪拌,傾溶液至蒸發皿中,置水浴上蒸去乙醇至約10 mL,移至分液漏斗中,用少量水洗滌蒸發皿,洗滌液倒入分液漏斗中,用乙醚振搖提取2次,每次15 mL,合并乙醚提取液,再向上述乙醚溶液中加2%碳酸鈉溶液振搖提取2次,每次15 mL,棄去乙醚液,提取液用鹽酸調節pH值至2~3,用少量苯洗滌,棄去苯液,再用乙醚振搖提取2次,每次30 mL,合并乙醚提取液,揮干,殘渣加乙醇1 mL溶解,作為供試品溶液。另取當歸和川芎對照藥材各1 g,分別加水100 mL,煎煮1 h,濾過,濾液濃縮至約10 mL,分別加乙醚振搖提取2次,每次15 mL,合并乙醚提取液,揮去乙醚,殘渣加乙醇1 mL使溶解,分別制成當歸和川芎的對照藥材溶液。另取阿魏酸對照品,加乙醇制成每1 mL含1 mg的溶液,作為對照品溶液。照薄層色譜法 (中國藥典2010年版一部附錄VI B)試驗,吸取上述各溶液各10 μL分別點于同一硅膠G薄層板上,以苯-冰醋酸-甲醇 (30∶1∶3)為展開劑,展開,取出,晾干,置紫外光燈 (365 nm)下檢視。供試品色譜中,在與對照藥材和對照品色譜相應的位置上,顯相同顏色的熒光斑點,陰性對照無干擾,見圖1。
2.2 白芍的薄層色譜鑒別[3]取當歸調經膠囊樣品10粒,研細,取粉末2 g,加水20 mL加熱溶解,置分液漏斗中,用水飽和的正丁醇提取2次,每次30 mL,合并正丁醇液,用水洗滌萃取液2次,每次20 mL。正丁醇液置水浴上濃縮至約1 mL,加中性氧化鋁0.5 g,拌勻,干燥,裝入中性氧化鋁柱 (100~200目,約1 g,內徑1~1.5 cm)上,以乙酸乙酯-甲醇 (1∶1)30 mL洗脫,收集洗脫液,蒸干,殘渣加乙醇1 mL使溶解,作為供試品溶液。另取白芍對照藥材的粉末1 g,加乙醇15 mL,振搖5 min,濾過,濾液蒸干,殘渣加乙醇1 mL使溶解,制成白芍的對照藥材溶液。另取芍藥苷對照品,加乙醇制成每1 mL含2 mg的溶液,作為對照品溶液。照薄層色譜法 (中國藥典2010年版一部附錄ⅤI B)試驗,吸取上述供試品溶液10 μL、對照品溶液5 μL,分別點于同一硅膠G薄層板上,以三氯甲烷-乙酸乙酯-甲醇-乙酸 (10∶4∶6∶0.8)為展開劑,展開,取出,晾干,噴5%香草醛硫酸溶液,熱風吹至斑點顯色清晰。供試品色譜中,在與對照品色譜相應的位置上,顯相同顏色的斑點,陰性對照液色譜中無干擾,見圖2。

圖1 當歸、川芎 (阿魏酸)的TLC色譜圖
3 HPLC測定當歸調經膠囊中阿魏酸
3.1 色譜條件[4-7]Kromasil-C18色譜柱(250 mm×4.6 mm,5 μm);流動相為甲醇-乙腈-1%冰乙酸溶液 (5∶1∶10);體積流量1.0 mL/min;柱溫為室溫;檢測波長為323 nm;進樣量10 μL;阿魏酸出峰時間為20 min;理論板數按阿魏酸計算應不低于2 000。
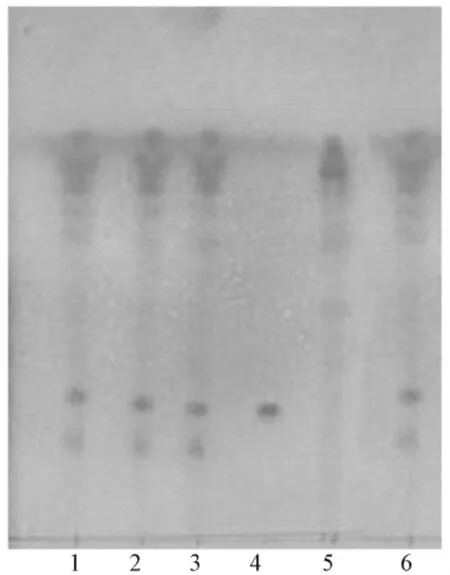
圖2 白芍 (芍藥苷)的TLC色譜圖
3.2 對照品溶液的制備 取阿魏酸對照品適量,精密稱定,加甲醇制成每1 mL含50 μg的溶液,即得。
3.3 供試品溶液的制備 取當歸調經膠囊樣品 (批號110201)10粒,研細,取粉末5 g,精密稱定,置分液漏斗中,用20 mL水溶解,加少量氯化鈉,用二氯甲烷提取4次 (每次20 mL),合并二氯甲烷層,用旋轉蒸發器揮干并回收二氯甲烷,殘渣用甲醇溶解并定容至5 mL量瓶中,作為供試品溶液。
3.4 陰性樣品溶液的制備 按當歸調經膠囊處方和制法,制成缺當歸和白芍陰性制劑,精密稱取5 g,再按供試品溶液制備方法制備陰性溶液。進樣20 μL測定,結果表明陰性樣品對阿魏酸測定無干擾,見圖3。
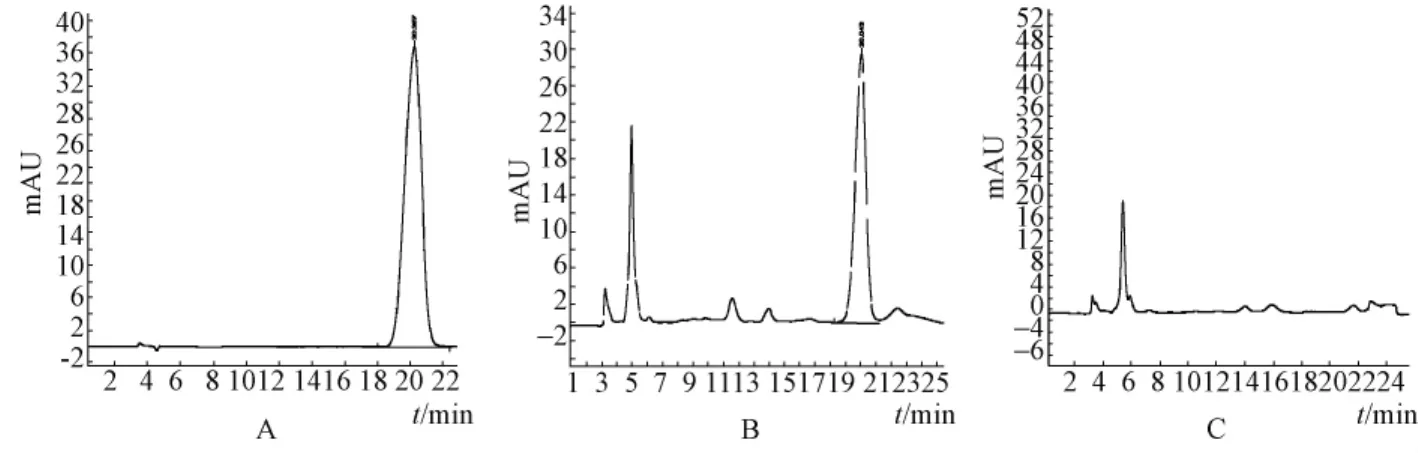
圖3 阿魏酸對照品 (A)、供試品 (B)及陰性樣品 (C)的HPLC色譜圖
3.5 標準曲線與線性范圍 精密稱取阿魏酸對照品7.68 mg,置25 mL量瓶中,加甲醇溶解并稀釋到刻度,作為貯備液 (0.307 2 mg/mL)。精密量取貯備液0.5、1、2、4、8 mL置10 mL量瓶中,加甲醇溶解并稀釋到刻度,分別精密吸取20 μL,注入液相色譜儀,按上述色譜條件測定。以阿魏酸的峰面積值為縱坐標,以質量濃度為橫坐標,進行線性回歸,得出回歸方程為:Y=5×107X,r=1,標準曲線為通過原點的一條直線,所以可以用外標一點法測定計算阿魏酸。結果表明阿魏酸在0.015 4~0.245 8 mg/mL質量濃度范圍內與峰面積線性關系良好。
3.6 精密度試驗 取同一對照品溶液 (質量濃度為51.2 μg/mL)及同一供試品樣品 (批號110201)溶液各重復進樣5次,每次20 μL,觀察其重復性。結果表明,對照品溶液阿魏酸峰面積的RSD為1.52%,供試品阿魏酸峰面積的RSD為0.93%,表明本法精密度好。
3.7 穩定性試驗 分別于0、2、4、6、8 h精密吸取阿魏酸對照品溶液 (質量濃度為51.2 μg/mL)及供試品溶液(批號110201)各20 μL注入液相色譜儀測定,對照品溶液峰面積的RSD為0.429%,供試品溶液峰面積的RSD為0.619%。結果表明,對照品及供試品溶液在8 h內穩定。
3.8 重復性試驗 取當歸調經膠囊 (批號110201)5份,分別精密稱定,按供試品溶液的制備方法制成供試品溶液,另取阿魏酸對照品適量,加甲醇制成1 mL含阿魏酸對照品0.048 mg的對照品溶液。
精密吸取供試品、對照品溶液各20 μL注入液相色譜儀,以甲醇-乙腈-1%冰乙酸溶液 (5∶1∶10)為流動相,測定,計算供試品溶液的RSD為1.83%,結果表明,方法重現性良好。
3.9 加樣回收率試驗 取已測定的當歸調經膠囊試制樣品3批 (批號:110201、110202、110203),各3份,約0.5 g,精密稱定,分別按其含有量的80%、100%、120%加入阿魏酸對照品適量,按供試品溶液的制備方法,制成供試品溶液,依法測定,計算回收率。結果平均回收率為98.10%,RSD為1.37%。見表1。
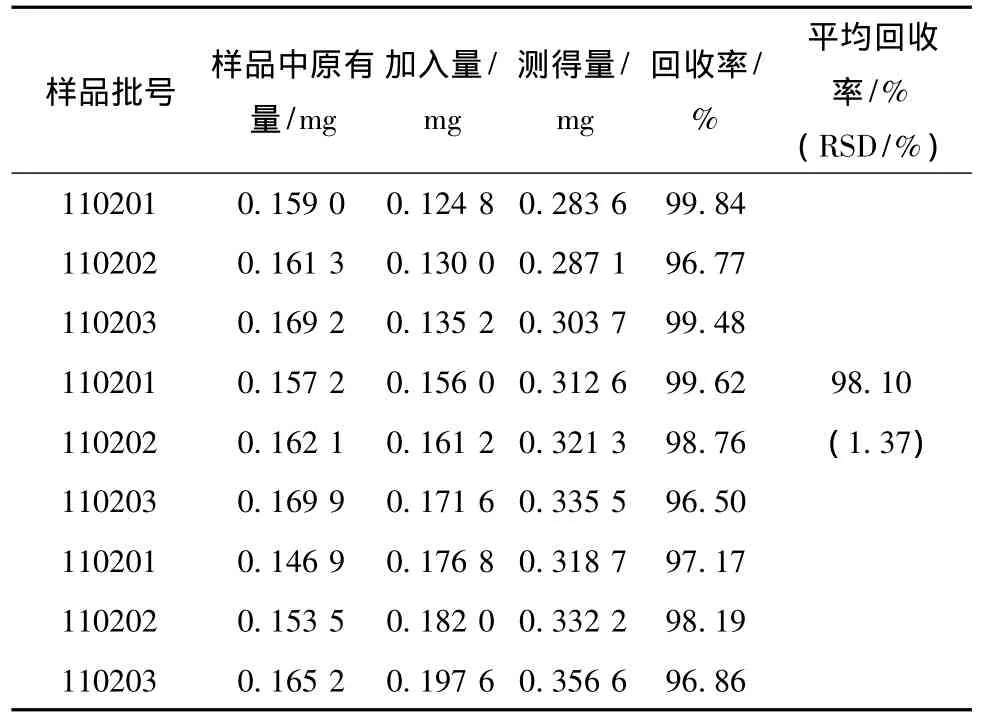
表1 阿魏酸加樣回收試驗結果
3.10 樣品測定 精密稱取3批當歸調經膠囊樣品 (批號:110201、110202、110203,每批制成3份供試品)各5 g,按供試品溶液的制備方法,制成供試品溶液,分別按3.1項下的色譜條件進行測定,結果見表2。
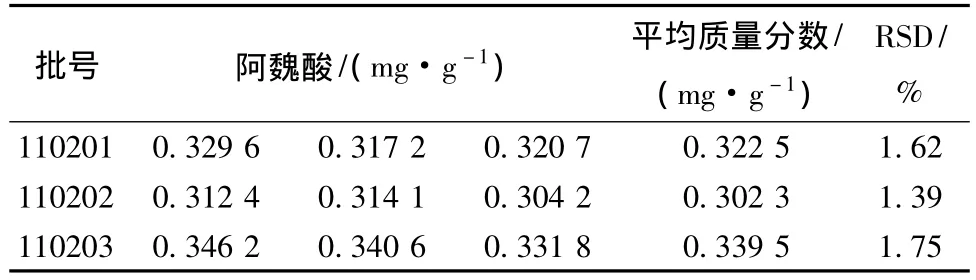
表2 樣品中阿魏酸測定結果 (n=3)
根據測定結果,每1 g樣品中阿魏酸量最高為:0.346 2 mg,最低為:0.304 2 mg,平均質量分數為0.324 1 mg/g,結合廠方工藝提取率以及藥材質量的差異,故本制劑阿魏酸含有量暫定為:每1 g樣品含阿魏酸 (C10H10O4)不得少于0.300 0 mg。
4 HPLC測定當歸調經膠囊中芍藥苷
4.1 色譜條件[8-11]Kromasil-C18色譜柱 (250 mm×4.6 mm,5 μm);流動相為乙腈-水 (18∶82);體積流量1.0 mL/min;柱溫為室溫;檢測波長為280 nm;進樣量10 μL;芍藥苷出峰時間為23 min;理論板數按阿魏酸計算應不低于2 000。
4.2 對照品溶液的制備 取芍藥苷對照品適量,精密稱定,加甲醇制成每1 mL含40 μg的溶液,即得。
4.3 供試品溶液的制備 取當歸調經膠囊樣品 (批號110201)10粒,研細,取粉末5 g,精密稱定,置具塞錐形瓶中,精密加入甲醇25 mL,密塞稱定質量。超聲處理25 min,放冷,再稱定質量,用甲醇補足減失質量,搖勻,濾過,取續濾液作為供試品溶液。
4.4 陰性樣品溶液的制備 按當歸調經膠囊處方和制法,制成缺白芍陰性制劑,精密稱取5 g,再按供試品溶液制備方法制備陰性溶液。進樣10 μL測定,結果表明陰性樣品對芍藥苷測定無干擾,見圖4。
4.5 標準曲線與線性范圍 精密稱取芍藥苷對照品8.02 mg,置20 mL量瓶中,加甲醇溶解并稀釋到刻度,作為貯備液 (0.401 mg/mL)。精密量取貯備液0.5、1、2、4、8 mL置10 mL量瓶中,加甲醇溶解并稀釋到刻度,分別精密吸取20 μL,注入液相色譜儀,按上述色譜條件測定。以芍藥苷的峰面積值為縱坐標,以質量濃度為橫坐標,進行線性回歸,得出回歸方程為:Y=2×107X,r=0.999 8,標準曲線為通過原點的一條直線,所以可以用外標一點法測定計算芍藥苷的量。結果表明芍藥苷在0.020 05~0.320 8 mg/mL質量濃度范圍內與峰面積線性關系良好。
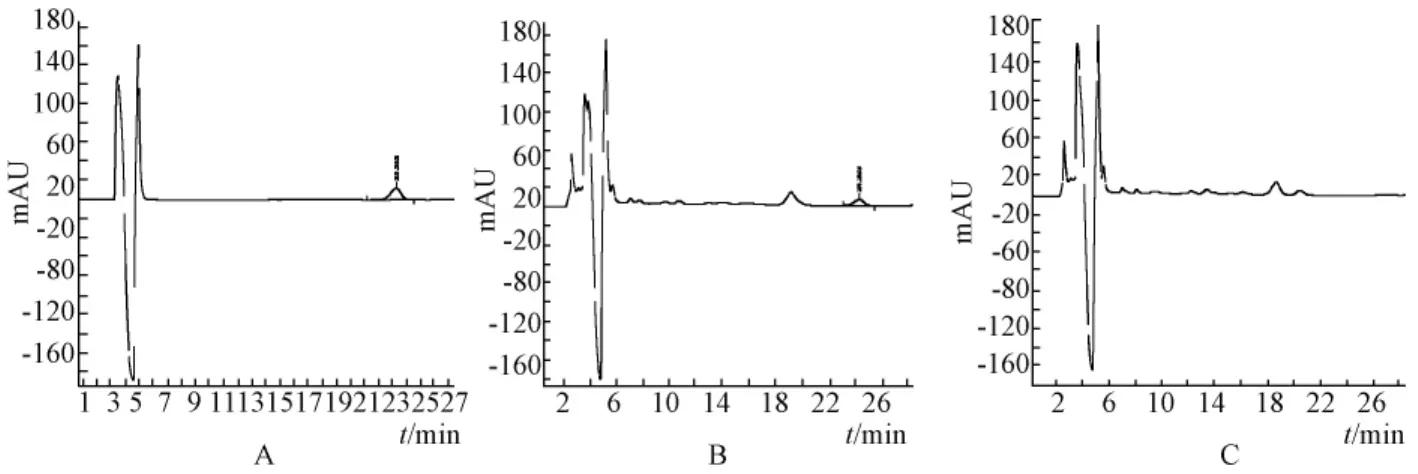
圖4 芍藥苷對照品 (A)、供試品 (B)及陰性樣品 (C)的HPLC色譜圖
4.6 精密度試驗 取同一對照品溶液 (質量濃度為40.1 μg/mL)及同一供樣品 (批號110201)溶液各重復進樣5次,每次20 μL,觀察其重復性。結果表明,對照品溶液芍藥苷峰面積的RSD為1.28%,供試品芍藥苷峰面積的RSD為1.53%,表明儀器精密度好。
4.7 穩定性試驗 分別于0、2、4、6、8 h精密吸取芍藥苷對照品溶液 (質量濃度為40.1 μg/mL)及供試品溶液(批號110201)各20 μL注入液相色譜儀測定,對照品溶液峰面積的RSD為1.68%,供試品溶液峰面積的RSD為1.75%。結果表明,對照品及供試品溶液在8 h內穩定。
4.8 重復性試驗 取當歸調經膠囊 (批號110201)5份,分別精密稱定,按供試品溶液的制備方法制成供試品溶液,另取芍藥苷對照品適量,加甲醇制成1 mL含芍藥苷對照品40 μg的對照品溶液。
精密吸取供試品、對照品溶液各20 μL注入液相色譜儀,測定,計算供試品中芍藥苷的量,其RSD為1.95%,結果表明,本法方法重復性良好。
4.9 加樣回收率試驗 取已測定的當歸調經膠囊試制樣品3批 (批號:110201、110202、110203),各3份,約2.5 g,精密稱定,分別按其含有量的80%、100%、120%加入芍藥苷對照品適量,按供試品溶液的制備方法,制成供試品溶液,依法測定,計算回收率。結果平均回收率為99.05%,RSD為1.02%。見表3。
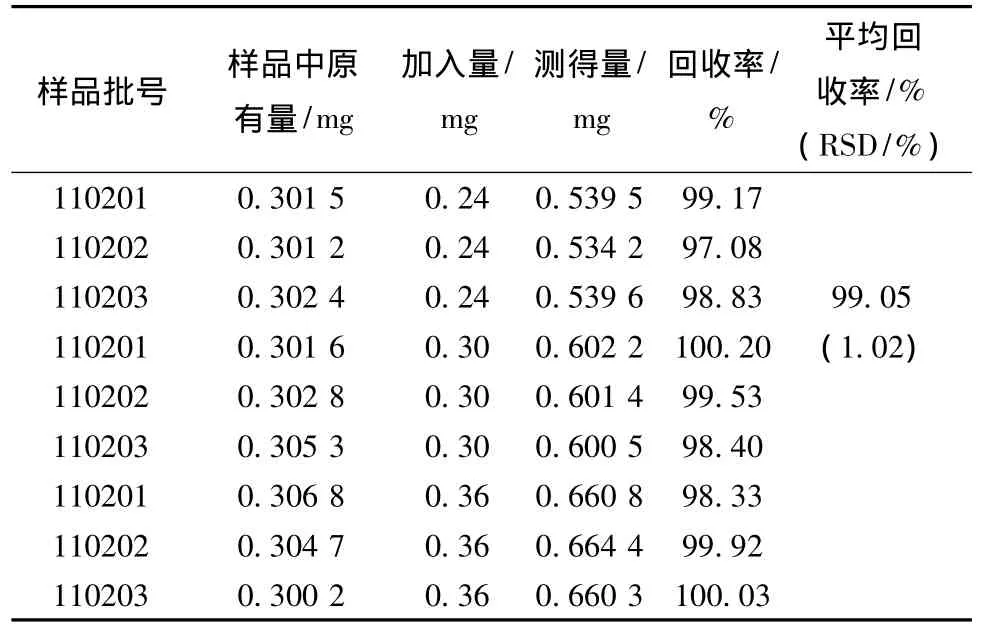
表3 芍藥苷加樣回收試驗結果
4.10 樣品測定 精密稱取3批當歸調經膠囊樣品 (批號:110201、110202、110203,每批制成3份供試品)各5 g,按供試品溶液的制備方法,制成供試品溶液,分別按3.1項下的色譜條件進行測定,結果見表4。
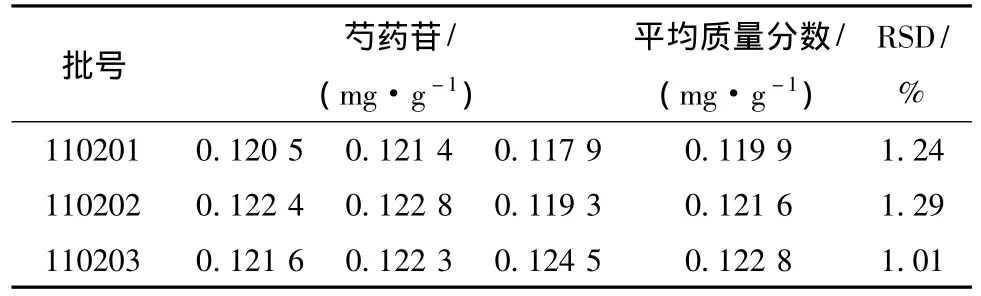
表4 樣品芍藥苷測定結果 (n=3)
根據測定結果,每1 g樣品中芍藥苷量最高為0.122 8 mg,最低為0.120 5 mg,平均質量分數為0.121 4 mg/g,結合廠方工藝提取率以及藥材質量的差異,故本制劑芍藥苷含有量暫定為每1 g樣品含芍藥苷不得少于0.120 0 mg。
5 討論
經反復實驗,通過優選實驗方法和展開劑,對當歸調經膠囊中當歸、川芎、白芍進行TLC鑒別,其色譜特征斑點清晰、專屬性強,所以此3味中藥可用于該制劑的定性鑒別。川芎、當歸均有活血調經、化瘀止痛的功效,其有效成分均為阿魏酸[12]、芍藥苷,對婦女月經不調有很強的藥理作用,所以阿魏酸、芍藥苷可以作為該制劑的指標性成分。當歸調經膠囊每1 g樣品中阿魏酸不得低于0.300 0 mg/g和芍藥苷不得低于0.120 0 mg,以此作為該制劑定量測定指標。
實驗選用甲醇-乙腈-1%冰乙酸溶液作流動相,增加甲醇量可改變峰形,減少拖尾,但甲醇比例過高,分離效果下降,不易與雜質峰分離。另外加入1%冰乙酸主要是調節流動相的pH,縮短分離時間,改善分離度。
[1]國家中醫藥管理局《中華本草》編委會.中華本草[M].上海:上海科學技術出版社,1999:893.
[2]姚慧娟,胡道德,顧 磊,等.靈杞黃斑膠囊的薄層鑒別研究[J].時珍國醫國藥,2009,20(11):2795-2796。
[3]黃艷萍,黃勇紅.當歸調經片的薄層鑒別和阿魏酸的測定[J].中國實驗方劑學雜志,2010,16(6):119-121.
[4]葉會呈,葉期馨,王帶媚,等.HPLC法測定血府逐瘀湯中阿魏酸的含量[J].中國新藥與臨床藥理,2009,20(4):356-358.
[5]王 健,范文成,葉曉紅,等.HPLC法測定婦康片中阿魏酸的含量[J].中國實驗方劑學雜志,2008,14(8):15-16.
[6]彭 紅,付建武,余日躍.復方當歸注射液中羥基紅花黃色素A和阿魏酸的含量測定[J].時珍國醫國藥,2009,20(12):2925-2927.
[7]譚曉虹,郭春燕,李 倩,等.桃紅四物湯水提液中沒食子酸、芍藥苷和紅花黃色素含量的測定[J].中成藥,2008,30(7):9945-997.
[8]毛曉敏,張小波,陳小清,等.HPLC同時測定十二烏雞白鳳丸中芍藥苷、阿魏酸和丹皮酚的含量[J].中成藥,2008,30(5):678-681.
[9]凌 明,馬安宇.HPLC測定胃腸合劑中芍藥苷和黃芩苷的含量[J].中成藥,2008,30(9):1311-1313.
[10]賀志偉,賀 慶,林云徑,等.RP-HPLC法測定杭白芍及其飲片中芍藥內酯苷、芍藥苷和苯甲酰芍藥苷[J].中草藥,2008,39(3):378-380.
[11]張葉萍,黃琴偉,龔 青,等.HPLC測定升血膏中芍藥苷的含量[J].中國中藥雜志,2008,33(2):200-201.
[12]呂光華,程世瓊,陳金泉,等.HPLC測定川芎藥材和飲片中游離阿魏酸和總阿魏酸的含量及其質量評價指標[J].中國中藥雜志,2010,35(2):194-197.
