靜電排斥表面誘導溶菌酶分子站立
2013-09-21 09:00:12白姝李浩張麟
物理化學學報 2013年4期
白 姝 李 浩 張 麟
(天津大學化工學院生物工程系,天津300072; 天津大學系統(tǒng)生物工程教育部重點實驗室,天津300072)
1 引言
基因工程技術的飛速發(fā)展推動重組蛋白質藥物在微生物體內的大規(guī)模生產.1-3但是,重組蛋白質在原核細胞中的高水平表達往往導致其發(fā)生錯誤的折疊和聚集,4形成被稱為包含體(inclusion body)的無活性聚集體,需要通過蛋白質復性過程使其恢復天然構型和生物學活性.5,6因此,蛋白質復性技術開發(fā)和理論完善對蛋白質藥物生產具有重要意義.7
蛋白質復性的最大挑戰(zhàn)就是創(chuàng)造合適的復性條件,最大程度地抑制聚集體生成,從而提高復性收率.8而現(xiàn)階段重組蛋白質復性仍存在收率低,操作條件較為嚴苛等問題.近期,Wang等9通過實驗研究發(fā)現(xiàn),帶有正電荷的離子交換介質Q和DEAE Sepharose FF可以顯著促進帶正電的溶菌酶復性,而帶有負電荷的離子交換介質SP和CM Sepharose FF可以顯著促進帶負電的牛血清白蛋白復性,并推斷與蛋白質帶有同種電荷的離子交換介質可以誘導蛋白質在介質表面形成定向排列.此定向排列促使蛋白質之間的靜電排斥力增大,從而抑制蛋白質的聚集而促進復性.但是,通過實驗研究很難直接證實此機理.而分子動力學(MD)模擬10,11能夠直接提供系統(tǒng)的微觀結構信息,并能通過數學統(tǒng)計等手段計算系統(tǒng)的宏觀性質,已經成為與理論研究和實驗研究并行互補的科學研究手段,廣泛用于蛋白質相關領域的研究.12迄今為止,已有大量研究者通過分子模擬研究界面吸引誘導的小分子13,14和蛋白質15-23吸附行為,但很少研究界面排斥作用誘導的蛋白質空間分布.
因此,本文將構建靜電排斥表面模型以模擬帶同種電荷的離子交換介質,通過分子動力學模擬考察蛋白質與靜電排斥表面之間的相互作用過程,展示蛋白質在靜電排斥表面上的空間取向和構象轉換,考察表面所帶電荷數的影響規(guī)律,揭示同電荷離子交換介質輔助蛋白質復性的微觀機理,推動蛋白質在荷電表面折疊和分子相互作用研究.
2 模擬方法
2.1 模擬體系構建
根據蛋白質數據庫(http://www.rcsb.org/pdb/)中的晶體衍射結構(PDB ID:3LYZ)構建溶菌酶的全原子模型,24如圖1A所示.溶菌酶由129個殘基組成,pH=7時帶8個正電荷(包括17個帶正電荷的殘基和9個帶負電荷的殘基).
本文構建靜電排斥表面模型以模擬同電荷離子交換介質,包括基質和配基兩部分(參比體系只含基質).基質由正六邊形網格結構的平板表示,其中正六邊形的邊長為0.152 nm.配基的原子組成參照商業(yè)化介質的化學結構(https://www.gelifesciences.com/)確定,其鍵長、鍵角和二面角及其力常數等結構參數由PRODRG 2.5(http://davapc1.bioch.dundee.ac.uk/cig-bin/prodrg_beta)25生成,而原子的部分電荷通過量子化學計算和類似結構比對調整確定,標示于圖1B中.其后,將配基連接到基質,根據實驗條件調整配基密度為0.22 mmol·mL-1,如圖1C所示.

圖1 溶菌酶和靜電排斥表面的全原子模型Fig.1 All-atom models of the lysozyme and the surface with electrostatic repulsion
構建模擬體系時,首先將靜電排斥表面放入6.84 nm×6.91 nm×18.00 nm的長方體盒子底部(參比體系的盒子高度為10.00 nm).然后將1個溶菌酶分子放置于表面上方,其質心距離表面約2 nm.進而添加水分子,并添加適量反離子使模擬體系呈電中性.最后將模擬體系放入6.84 nm×6.91 nm×100 nm的長方體盒子中心進行模擬.本文共構建4個模擬體系,包括靜電排斥表面體系(標記為Q),參比體系(標記為control),介質所帶電荷增大30%(標記為1.3Q)和減小25%(標記為0.75Q,體系高度為12 nm)的體系,其具體組成列于表1.
2.2 分子動力學模擬
分子動力學模擬利用GROMACS 4.0.5程序(http://www.gromacs.org/)26,27完成,使用GROMOS96 43A1力場.28模擬采用NVT系綜,通過v-rescale溫度偶聯(lián)法29控制溫度為298.15 K.采用半步蛙跳法積分,步長為2 fs.靜電相互作用計算采用PME(particle-mesh Ewald)算法,其截斷距離設為1.2 nm.范德華作用計算采用cut-off算法,其截斷半徑設為1.2 nm.近鄰原子列表截斷半徑設為1.2 nm.粒子運動的初始速度根據模擬溫度298.15 K下的Maxwell分布產生.對模擬盒子施加x,y,z的三個方向的周期性邊界條件.
本文采用最速下降法進行能量最小化,能量閥值設置為2000 kJ·mol-1.然后,進行20 ns的分子動力學模擬,得到模擬軌跡用于后續(xù)分析.為了保證模擬結果的準確性,本文中對體系Q執(zhí)行3次平行模擬.所有模擬均使用曙光TC2600刀片服務器完成.文中蛋白質構象圖利用RASMOL軟件30繪制.
2.3 數據分析方法
2.3.1 蛋白質空間取向分析
本文定義角度θ為蛋白質的偶極和z坐標軸之間的夾角,以定量描述蛋白質在靜電排斥表面上的空間取向.當θ=0°時,蛋白質的偶極垂直于靜電排斥表面(平行于+z坐標軸).當θ為90°或-90°時,蛋白質的偶極平行于靜電排斥表面(偶極指向+x方向定義為正).通過自編程序計算蛋白質的θ值隨模擬時間的變化.

表1 模擬體系的參數Table 1 Parameters of simulation systems
2.3.2 分子間勢能分析
利用GROMACS軟件包的g_energy程序進行勢能分析,包括Lennard-Jones(LJ)勢能(標記為ELJ)和庫侖勢能(標記為EC).
2.3.3 二級結構分析
為了描述MD模擬過程中蛋白質的構象轉化,利用Define Secondary Structure of Proteins(DSSP)方法31及GROMACS軟件包的do_dssp程序完成其二級結構分析.
2.3.4 均方根偏差計算
利用GROMACS軟件包的g_rms程序計算溶菌酶分子相比于其天然結構的均方根偏差(RMSD),如式(1)所示.RMSD值越小,表明溶菌酶天然結構保持更好.
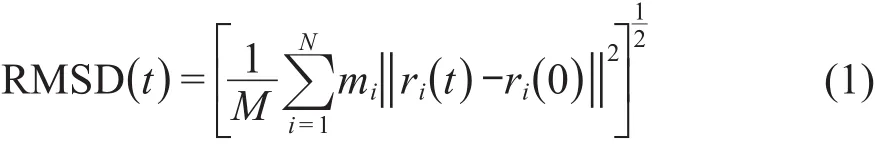
2.3.5 回轉半徑計算
利用GROMACS軟件包的g_gyrate程序計算溶菌酶的回轉半徑(Rg)以描述其結構緊密程度,如式(2)所示.Rg值越小,表明蛋白質的結構越緊密.
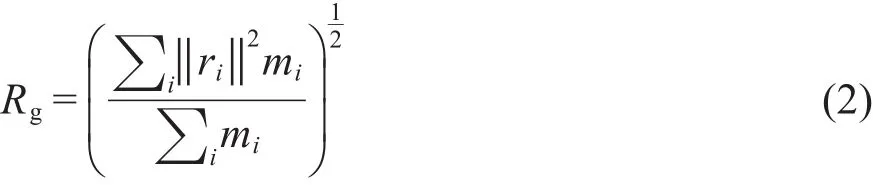
2.3.6 自由能表面圖
參照文獻20,32計算蛋白質處于特定θ和zcom的概率分布P(θ,zcom),其中zcom為蛋白質質心的z坐標值.通過模擬軌跡采樣和自編程序計算完成.
3 結果與討論
3.1 溶菌酶空間取向分析
首先通過系統(tǒng)快照(snapshots)直觀分析模擬體系的微觀構象隨模擬時間的變化,包括溶菌酶的構象,空間取向以及位置等,如圖2所示.此處,著重分析溶菌酶的偶極取向,偶極已放大以便清晰顯示,在圖中以棒狀標出,其偶極方向由紅色指向藍色(印刷版中由黑色指向灰色).注意,此處僅顯示一條典型軌跡結果,其余平行模擬軌跡的分析結果一致,故略去.
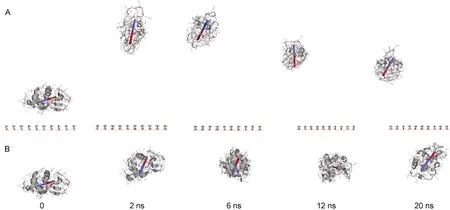
圖2 溶菌酶在靜電排斥表面上的構象隨模擬時間變化圖Fig.2 Snapshots of lysozyme over the surface with electrostatic repulsion as a function of simulation time
圖2A顯示,溶菌酶的初始位置靠近靜電排斥表面,其偶極方向接近平行方向,溶菌酶含有豐富的α-螺旋和β-折疊等二級結構.其后,蛋白質受到表面的排斥作用而遠離.2 ns時,蛋白質已經遠離表面并呈現(xiàn)“站立”姿勢,其偶極方向接近垂直方向.此時蛋白質整體結構被拉伸,其二級結構部分喪失而轉變?yōu)橄鄬θ嵝缘木砬Y構.6 ns時,蛋白質依然遠離表面,其偶極方向有輕微偏轉,但依然接近垂直方向,即蛋白質一直保持“站立”姿勢.其后,由于蛋白質遠離表面,且偶極旋轉會進一步降低表面對其的排斥作用,蛋白質可以再次靠近表面.例如,12 ns時,蛋白質略微靠近表面,而其偶極旋轉到幾乎完全垂直的方向,表明蛋白質靠近表面時,靜電相互作用增強利于誘導形成“站立”姿勢.但同時也需注意,20 ns時,蛋白質進一步接近表面,但其偶極反而偏離垂直方向,表明蛋白質的取向還受到其他因素,如溶劑的影響,存在一定波動.
在參比體系(圖2B)中,由于表面呈電中性,和蛋白質之間不存在靜電相互作用.因此,蛋白質一直保持在表面附近,在溶劑作用下存在一定的移動和轉動,但其偶極方向變化并無明顯規(guī)律.同時,在參比體系中,蛋白質的二級結構一直很好保持,表明蛋白質自身結構在溶液中比較穩(wěn)定.
因此,微觀構象分析表明,靜電排斥表面推動蛋白質遠離.在此過程中,表面誘導蛋白質逐漸“站立”,使其偶極保持在垂直于表面的方向.盡管蛋白質的偶極方向存在一定的擺動,但總體而言都接近垂直方向,即蛋白質基本保持“站立”姿勢.
進而考察角度θ(定義和計算流程見2.3.1節(jié))的變化以定量描述蛋白質在靜電排斥表面作用下的空間取向分布及其變化情況,同時通過蛋白質質心的z坐標值定量描述蛋白質與表面的相對位置變化,如圖3所示.此處,體系Q的數據為3次平行模擬結果的平均值.
在靜電排斥表面上,蛋白質的θ值從初始的-105°迅速變化到0°左右,即蛋白質從“平躺”姿勢迅速轉變?yōu)椤罢玖ⅰ弊藙?同時其zcom迅速增大,表明蛋白質迅速遠離表面.相比而言,zcom變化更緩慢,即其變化速率小于θ的變化速率,表明蛋白質在受到排斥而遠離表面的過程中即已形成“站立”姿勢.在2 ns附近時,蛋白質的zcom呈現(xiàn)峰值,此時蛋白質的θ在0°附近波動,但都維持在-60°到60°的區(qū)間內,表明蛋白質在遠離表面時,由于靜電排斥減弱,蛋白質的偶極方向存在一定的擺動.其后,蛋白質的zcom值略微降低,在3.0到5.5 ns期間呈現(xiàn)平臺期,其zcom值維持在8 nm,而此時蛋白質的θ值基本維持在0°,表明蛋白質依然很好維持“站立”姿勢.此后,蛋白質略微接近表面,而θ值的變化幅度增大,但依然維持在0°附近.到12 ns后,蛋白質的θ值變化幅度進一步增大,表明蛋白質從遠離靜電排斥表面的位置逐漸靠近表面時,由于靜電排斥作用增強,蛋白質的偶極方向需要重新調整以達到新的平衡狀態(tài).其概率分布(圖3B)表明,蛋白質的zcom主要介于5到10 nm之間,表明蛋白質受到排斥而遠離表面,其θ角主要分布于-60°到60°之間,確證蛋白質形成“站立”姿勢.

圖3 溶菌酶空間取向和位置隨模擬時間變化圖Fig.3 Orientation and the location of lysozyme as a function of simulation time
在參比體系中,蛋白質一直維持在表面附近,其zcom值變化較小,可以認為蛋白質只是在其初始位置附近波動而并無明顯移動(圖3C的統(tǒng)計結果給出明確證據).在模擬初始階段,蛋白質的θ值也無明顯變化,只是在其初始值附近波動.2.5 ns后蛋白質的θ值發(fā)生明顯變化,但分析表明都是無規(guī)的轉動導致,從圖2的系統(tǒng)快照中也可得到直觀證明.15 ns后,蛋白質的偶極方向又恢復到初始狀態(tài)并存在輕微波動.
因此,θ值和zcom值隨模擬時間的變化確證蛋白質在靜電排斥表面作用下迅速遠離,而在遠離過程中其偶極旋轉,形成特定的“站立”姿勢.此結果直接驗證實驗推測,表明蛋白質確實在靜電排斥表面形成定向排布,有助于增強蛋白質之間的靜電排斥作用而抑制溶菌酶聚集.但同時需注意,蛋白質和表面間的距離,以及溶劑均影響蛋白質的偶極方向使其存在一定的擺動.
然后,通過蛋白質和表面間相互作用勢能分析,考察蛋白質形成特定“站立”姿勢的關鍵作用力,如圖4所示.此處,體系Q的數據為3次平行模擬結果的平均值.

圖4 溶菌酶和介質間的LJ勢能和庫侖勢能隨模擬時間變化圖Fig.4 Lennard-Jones(LJ)and Coulomb potential energies between lysozyme and resin as a function of simulation time
僅在模擬初始階段觀察到靜電排斥表面和蛋白質之間存在很小的LJ勢能,約-1.2 kJ·mol-1.其后,LJ勢能持續(xù)為0,這是因為蛋白質被排斥遠離表面(圖3)而LJ勢能是短程相互作用,隨距離迅速衰減.而蛋白質和表面之間存在很大的靜電相互作用勢能,接近140 MJ·mol-1.靜電相互作用勢能隨模擬時間迅速減小,到2 ns附近時達到最小值83 MJ·mol-1,這是因為蛋白質在靜電排斥作用下迅速遠離表面(圖3),使靜電勢能迅速降低.其后,隨著蛋白質再次靠近表面(圖3),蛋白質和表面間的靜電相互作用勢能再次上升,最終達到105 MJ·mol-1.而在參比體系中,蛋白質與表面之間的LJ勢能和靜電作用勢能都基本為0,因而表面對蛋白質并無作用,蛋白質呈現(xiàn)溶液主體行為,與圖2和圖3的結果完全吻合.
因此,通過勢能分析可以確證,表面通過靜電排斥作用驅動蛋白質遠離并誘導其形成特定“站立”姿勢.為驗證此結論,本文還構建帶負電的靜電表面,發(fā)現(xiàn)帶正電的溶菌酶分子在靜電吸引作用下直接吸附于負電表面(數據未顯示).如果通過調整溶液pH值使溶菌酶也帶負電(如pH=14),則溶菌酶同樣在靜電排斥作用下迅速遠離,再次確證靜電排斥作用是關鍵驅動力.但同時也發(fā)現(xiàn),pH=14時,溶菌酶中只有帶負電的氨基酸殘基而無帶正電的氨基酸殘基,無法形成明顯偶極而導致無法形成特定“站立”姿勢(數據未顯示),表明正負電荷的不均勻分布是特定“站立”姿勢形成的關鍵因素.
3.2 溶菌酶構象分析
利用DSSP方法確定蛋白質二級結構隨模擬時間的變化,如圖5所示.結果表明,溶菌酶的主要二級結構是α-螺旋,間雜零星β-折疊結構.參比體系中,各顏色條帶隨模擬進行都比較穩(wěn)定.例如,10號殘基、30號殘基和95號殘基處的α-螺旋在模擬過程中一直保持完好,而110號殘基處的α-螺旋結構存在短暫消失的情況,但很快重新生成,且一直維持到模擬終點,表明蛋白質的二級結構得到很好保持.
但在靜電排斥表面體系中,各顏色條帶均存在消失或增加的反復過程,總體呈現(xiàn)不連續(xù)分布狀態(tài),表明蛋白質的二級結構存在明顯變化.其變化細節(jié)表明蛋白質中多處的α-螺旋結構喪失.參照圖2的系統(tǒng)快照分析,在靜電排斥表面作用下,蛋白質中帶正電荷的殘基受到排斥而試圖遠離表面,但帶負電荷的殘基受到吸引而試圖靠近表面,因此蛋白質受力不均而被輕微拉伸,其內部原子相對位置的變化造成其二級結構的破壞.從圖5中也可看出,帶電殘基處的二級結構變化最為劇烈.例如,天然溶菌酶中ARG5-LYS13形成α-螺旋結構,在參比體系中此結構很好保持(圖5B).而在靜電排斥表面體系中,由于ARG5和LYS13均帶電,受到表面的影響而導致此螺旋結構不穩(wěn)定,甚至整體螺旋結構被破壞(圖5A).同時需注意,隨著蛋白質位置和偶極方向的調整,蛋白質構象也存在細微調整過程.
因此,計算蛋白質的RMSD和Rg值模擬時間的變化,以通過結構偏差程度及其整體結構緊密程度(詳見2.3節(jié)中數據分析方法描述)定量考察蛋白質的構象變化,如圖6所示.此處數據為3次平行模擬結果的平均值.
參比體系中,蛋白質的RMSD值在模擬開始時輕微增大,表明蛋白質在溶液環(huán)境中存在細微結構調整,其后則一直在0.10 nm上下波動.蛋白質的Rg值隨模擬時間輕微降低,然后維持在1.35 nm上下波動.二者的數值都很小且波動幅度也很小,表明參比體系中蛋白質并無明顯構象變化,與圖2的系統(tǒng)快照和圖5的二級結構分析結果一致.
而在靜電排斥表面體系中,蛋白質的RMSD值在模擬初始階段即迅速增大,并進而維持在0.5 nm上下波動且持續(xù)到模擬終點,表明蛋白質確實在模擬初始的排斥遠離過程中受到很大的結構擾動,使其結構明顯偏離天然態(tài).蛋白質的Rg值在模擬初始階段也迅速上升,高于參比體系,表明蛋白質結構確實被拉伸而使其整體結構松散.其后,Rg值很快下降到與參比體系相當,進而繼續(xù)下降并穩(wěn)定在1.30 nm上下波動,其穩(wěn)定值低于參比體系中的Rg值.綜合RMSD值和Rg值的變化表明蛋白質被排斥遠離表面后,由于距離增大(圖3)導致靜電排斥力減弱(圖4),蛋白質受到外界干擾減小,開始構象調整,呈現(xiàn)整體塌縮的變化趨勢.然而,蛋白質的最終結構仍然偏離天然結構,但其偏差數值并不大,表明其最終骨架結構還能較好保持,與圖2的系統(tǒng)快照分析結果一致.因而,靜電排斥表面對溶菌酶的分子結構有一定擾動,盡管整體骨架結構變化不大,也可能造成其活性喪失.
因此,綜合分析二級結構,RMSD和Rg值的結果表明,參比體系中,蛋白質的構象很好保持.而在靜電排斥表面體系中,蛋白質由于受力不均,存在先拉伸后塌縮的變化過程,其二級結構遭到破壞,但主體骨架結構較好保持.
3.3 表面所帶電荷數的影響分析
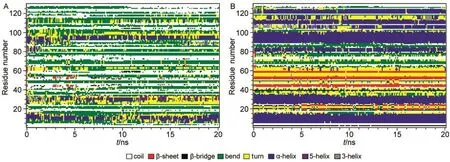
圖5 分子動力學模擬中溶菌酶分子的二級結構演化Fig.5 Secondary structure evolution of lysozyme during the molecular dynamics simulation
上述結果表明,靜電排斥表面能夠誘導蛋白質形成“站立”姿勢,而靜電排斥作用是主要驅動力.因此,表面所帶電荷數對蛋白質的空間取向應該具有重要影響.本文構建電荷數增大的靜電排斥表面模型(1.3Q)以及電荷數減小(0.75Q)的模型,分析其與蛋白質的相互作用及其對蛋白質的空間取向和構象變化的影響,如圖7、8、9所示.
圖7顯示,在體系1.3Q中,蛋白質的θ值一直維持在0°上下波動,且波動幅度很小,表明表面所帶電荷數增大使其靜電排斥作用增強,蛋白質能夠更好形成并保持“站立”姿勢.蛋白質的zcom值迅速增大,到6 nm處增大變緩,最終維持在10 nm上下波動.其概率分布(圖7B)表明,蛋白質的θ角更集中于-60°到60°之間.因此,相比于體系Q(圖3),蛋白質的排斥遠離過程更快且最終穩(wěn)定距離更遠,但其“站立”姿勢也更穩(wěn)定,表明表面所帶電荷數增大確實有利于蛋白質的“站立”姿勢形成和穩(wěn)定.
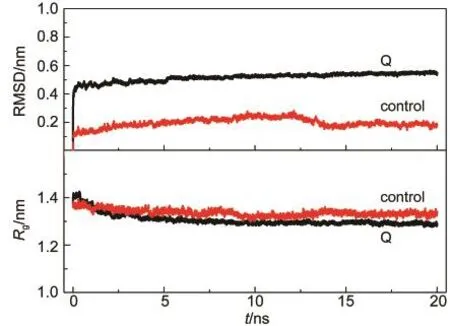
圖6 溶菌酶中Cα原子的RMSD和Rg隨模擬時間變化圖Fig.6 RMSD and the Rgvalues for Cαatoms of lysozyme as a function of simulation time
而在體系0.75Q中,蛋白質的θ值主要維持在-50°上下波動且波動幅度很大.在17至20 ns期間,蛋白質遠離表面,其θ值發(fā)生明顯變化,在-120°至-60°區(qū)間內波動,即偶極平行于表面,蛋白質呈現(xiàn)“平躺”姿勢.蛋白質的zcom值到1.6 ns時增大到5.7 nm,變化比體系1.3Q和體系Q中緩慢.其后,zcom值降低,到5 ns時再次上升,最終達到7.2 nm.因此,體系0.75Q中,模擬終點時zcom值小于體系1.3Q的情況,表明靜電排斥減弱,蛋白質穩(wěn)定位置更靠近表面.但需要注意的是,雖然體系0.75Q中模擬終點的zcom值大于體系Q,但其概率分布(圖7C)表明,在zcom值較大區(qū)域蛋白質的θ角偏離-60°到60°區(qū)域,表明蛋白質此時并不存在“站立”姿勢.而其主要分布區(qū)域是在zcom=5 nm處,此時其θ=-60°,表明體系0.75Q中,蛋白質也形成“站立”取向,但趨勢更弱.
圖8顯示,在1.3Q和0.75Q兩體系中,蛋白質和表面之間的LJ勢能都基本為0,與體系Q相同,表明在靜電排斥表面體系中,蛋白質和表面間距離較遠,而LJ勢能為短程相互作用,隨距離增大迅速衰減.而靜電相互作用很強,在兩體系中均呈現(xiàn)迅速下降,然后趨于平穩(wěn)的變化趨勢,分別穩(wěn)定在83和19 MJ·mol-1.勢能變化表明蛋白質的位置和空間取向變化受靜電排斥作用調控,且受到表面所帶電荷數影響.表面電荷數增大,蛋白質的平衡位置更遠離表面,且利于“站立”姿勢的形成和維持.
圖9顯示,在1.3Q和0.75Q兩體系中,蛋白質的RMSD值均隨模擬進行而迅速上升,然后分別在0.70和0.15 nm上下波動.而兩體系中蛋白質的Rg值略微不同,體系1.3Q中Rg值高于初始天然態(tài),且存在顯著增大再回落的過程(如2.6 ns時).而體系0.75Q中Rg值基本保持在天然態(tài)的Rg值.結合RMSD和Rg值分析表明,隨著所帶電荷數增大,表面對蛋白質的結構擾動加劇.反之,電荷數降低的表面對蛋白質的結構擾動減弱,使蛋白質能夠更好保持其天然結構.
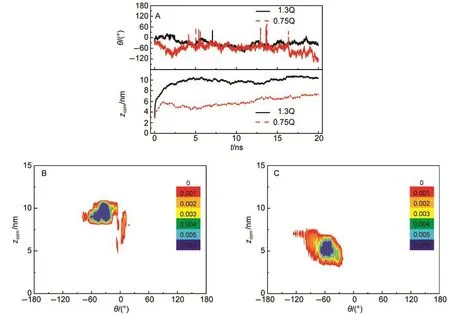
圖7 介質電荷數對溶菌酶的空間取向和位置的影響Fig.7 Effects of charge number of resin on the orientation and location of lysozyme
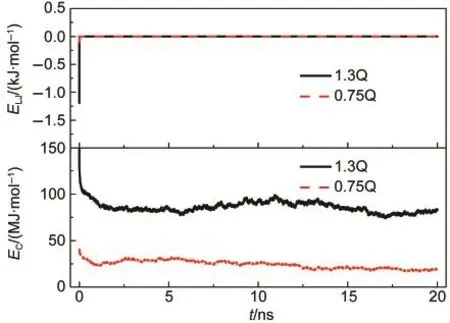
圖8 介質電荷數對溶菌酶和介質間相互作用勢能的影響Fig.8 Effects of charge number of resin on the interaction energies between the lysozyme and the resin
4 結論

圖9 介質電荷數對溶菌酶構象的影響Fig.9 Effects of charge number of resin on the conformation of lysozyme
以同電荷離子交換介質促進溶菌酶等蛋白質復性的實驗結果為背景,通過靜電排斥表面模擬同電荷離子交換介質,構建溶菌酶在靜電排斥表面體系中的全原子模型,利用分子動力學模擬考察蛋白質在表面上的空間取向和構象變化,并通過相互作用勢能分析深入剖析靜電排斥表面的作用機理和細節(jié).結果表明,蛋白質在表面的靜電排斥作用下迅速遠離,在此過程中逐漸形成其偶極垂直于表面的“站立”姿勢.此定向排布有助于增強蛋白質之間的靜電排斥作用而抑制溶菌酶聚集.但是,由于蛋白質中帶正電荷的殘基和帶負電荷的殘基受力方向相反,導致蛋白質結構拉伸而破壞其二級結構.研究發(fā)現(xiàn)表面所帶電荷數對蛋白質的空間取向和構象變化有顯著影響.電荷數增大使得表面對蛋白質的靜電排斥作用增強,能夠誘導蛋白質更快更好地形成“站立”姿勢.而表面所帶電荷數降低,則不利于蛋白質維持“站立”姿勢.同時,本文的模擬中尚有四個影響因素需要在后續(xù)研究中考慮.其一,模擬體系中的大量溶劑分子會影響蛋白質的構象和空間取向調整.其二,本文研究的排斥性界面不同于吸附性界面.吸附性界面作用下,蛋白質靠近界面使得相互作用增強,因而利于形成明顯的空間取向分布.而排斥性界面作用下,蛋白質遠離界面,使得相互作用減弱,不利于蛋白質形成明顯空間取向,造成排斥性界面研究的困難.其三,限于計算量和模擬體系復雜度的考慮,本文僅模擬單個溶菌酶分子,能夠確證靜電排斥表面誘導蛋白質形成定向排布,但無法直接闡釋表面對蛋白質分子間相互作用的影響,將在后續(xù)研究中開展.其四,本文直接用晶體結構建模計算,后續(xù)研究中可以先通過一定時間的模擬獲得溶菌酶在“自由”狀態(tài)下的構象以消除溶液條件和力場的影響.總體而言,本文通過分子動力學模擬完整闡釋蛋白質在靜電排斥表面上的空間取向和構象變化過程,考察此過程的微觀細節(jié),并能確定表面電荷數等參數的影響規(guī)律,將有助于推動蛋白質在荷電表面折疊和分子相互作用研究.
(1) Durocher,Y.;Butler,M.Curr.Opin.Biotech.2009,20,700.doi:10.1016/j.copbio.2009.10.008
(2)Manning,M.C.;Chou,D.K.;Murphy,B.M.;Payne,R.W.;Katayama,D.S.Pharm.Res.2010,27,544.doi:10.1007/s11095-009-0045-6
(3) Ferrer-Miralles,N.;Domingo-Espin,J.;Corchero,J.L.;Vazquez,E.;Villaverde,A.Microb.Cell.Fact.2009,8,17.doi:10.1186/1475-2859-8-17
(4) Fink,A.L.Fold.Des.1998,3,R9.
(5) Middelberg,A.R.Trends Biotechnol.2002,20,437.doi:10.1016/S0167-7799(02)02047-4
(6) Baldwin,R.L.J.Mol.Biol.2007,371,283.doi:10.1016/j.jmb.2007.05.078
(7)Chen,Y.W.;Ding,F(xiàn).;Nie,H.F.;Serohijos,A.W.;Sharma,S.;Wilcox,K.C.;Yin,S.Y.;Dokholyan,N.V.Arch.Biochem.Biophys.2008,469,4.doi:10.1016/j.abb.2007.05.014
(8) Lu,D.;Liu,Z.Annu.Rep.Prog.Chem.,Sect.C 2010,106,259.doi:10.1039/b903487k
(9)Wang,G.;Dong,X.;Sun,Y.Biotechnol.Bioeng.2011,108,1068.doi:10.1002/bit.23038
(10) Karplus,M.Biopolymers 2003,68,350.
(11) Karplus,M.;McCammon,J.A.Nat.Struct.Biol.2002,9,646.doi:10.1038/nsb0902-646
(12) Bai,H.J.;Lai,L.H.Acta Phys.-Chim.Sin.2010,26,1988.[白紅軍,來魯華.物理化學學報,2010,26,1988.]doi:10.3866/PKU.WHXB20100725
(13)Zhang,X.R.;Wang,W.C.Acta Phys.-Chim.Sin.2002,18,680.[張現(xiàn)仁,汪文川.物理化學學報,2002,18,680.]doi:10.3866/PKU.WHXB20020803
(14) Xiang,Z.H.;Wang,W.C.;Cao,D.P.Scientia Sinica Chimica 2012,42,235.[向中華,汪文川,曹達鵬.中國科學:化學,2012,42,235.]doi:10.1360/032011-297
(15) Kubiak-Ossowska,K.;Mulheran,P.A.Langmuir 2010,26,15954.doi:10.1021/la102960m
(16) Ravichandran,S.;Madura,J.D.;Talbot,J.J.Phys.Chem.B 2001,105,3610.doi:10.1021/jp010223r
(17) Carlsson,F(xiàn).;Hyltner,E.;Arnebrant,T.;Malmsten,M.;Linse,P.J.Phys.Chem.B 2004,108,9871.doi:10.1021/jp0495186
(18)Slater,G.W.;Holm,C.;Chubynsky,M.V.;de Haan,H.H.;Dube,A.;Grass,K.;Hickey,O.A.;Kingsburry,C.;Sean,D.;Shendruk,T.N.;Nhan,L.X.Electrophoresis 2009,30,792.doi:10.1002/elps.v30:5
(19) Chandrasekhar,I.;Kastenholz,M.;Lins,R.D.;Oostenbrink,C.;Schuler,L.D.;Tieleman,D.P.;van Gunsteren,W.F.Eur.Biophys.J.Biophy.2003,32,67.
(20) Zhang,L.;Zhao,G.F.;Sun,Y.J.Phys.Chem.B 2009,113,6873.doi:10.1021/jp809754k
(21) Zhang,L.;Zhao,G.F.;Sun,Y.J.Phys.Chem.B 2010,114,2203.doi:10.1021/jp903852c
(22)Kang,K.;Lu,D.N.;Liu,Z.Chin.J.Chem.Eng.2012,20,284.
(23) Shen,L.M.;Lin,D.Q.;Mei,L.H.;Yao,S.J.Chinese Journal of Bioprocess Engineering 2006,4,29.[沈立民,林東強,梅樂和,姚善涇.生物加工過程,2006,4,29.]
(24) Diamond,R.J.Mol.Biol.1974,82,371.doi:10.1016/0022-2836(74)90598-1
(25)Schuttelkopf,A.W.;vanAaltern,D.M.F.Acta Crystallogr.D 2004,60,1355.doi:10.1107/S0907444904011679
(26) Berendsen,H.J.;van der Spoel,D.;van Drunen,R.Comput.Phys.Commun.1995,91,43.doi:10.1016/0010-4655(95)00042-E
(27) Lindahl,E.;Hess,B.;van der Spoel,D.J.Mol.Model.2001,7,306.
(28)Mackerell,A.D.J.Comput.Chem.2004,25,1584.
(29) Bussi,G.;Donadio,D.;Parrinello,M.J.Chem.Phys.2007,126,014101.doi:10.1063/1.2408420
(30) Sayle,R.;Milnerwhite,E.Trends Biochem.Sci.1995,20,374.doi:10.1016/S0968-0004(00)89080-5
(31) Kabsch,W.;Sander,C.Biopolymers 1983,22,2577.
(32)Li,W.F.;Zhang,J.;Wang,J.;Wang,W.J.Am.Chem.Soc.2008,130,892.doi:10.1021/ja075302g
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中華詩詞(2019年7期)2019-11-25 01:43:04
中國外匯(2019年17期)2019-11-16 09:31:14
模具制造(2019年3期)2019-06-06 02:10:54
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
現(xiàn)代企業(yè)(2015年9期)2015-02-28 18:56:50
現(xiàn)代企業(yè)(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
土木建筑工程信息技術(2013年2期)2013-10-17 03:14:12
