活性炭基軟包裝超級電容器用有機電解液
2013-09-21 09:00:20孫現眾張大成馬衍偉
物理化學學報 2013年9期
黃 博 孫現眾 張 熊 張大成 馬衍偉
(中國科學院電工研究所應用超導重點實驗室,北京100190)
1 引言
超級電容器(supercapacitors或ultracapacitors)也叫電化學電容器,1?4因其具有適中的能量密度和功率密度、較長的循環壽命、綠色環保等優點,在新能源發電、電動汽車、信息技術、航空航天和國防等領域都有著廣闊的應用前景.5?10超級電容器電極材料主要分為碳材料、金屬氧化物、導電聚合物等.目前,在很多文獻中,超級電容器專指以碳材料為電極材料的電化學雙電層電容器,本文亦沿用這一用法.超級電容器靠活性物質與電解液的界面存儲電荷,11沒有法拉第反應發生,12其存儲的能量E=1/2·CU2.由此可知,電容器的能量密度E與比電容C和工作電壓U的平方均成正比.4因此可以從提高電極材料比電容和工作電壓兩方面入手來提高超級電容器的比能量.13超級電容器的比電容往往由電極材料和電解液共同決定,14而電壓窗口主要由電解液決定.因此,電解液對超級電容器的比能量有著決定性的作用.
目前所使用的超級電容器電解液有水系電解液、有機電解液和離子液體.水系電解液多為離子導電性較好的硫酸、氫氧化鉀等溶液,比電容高但電壓窗口窄(只有1 V左右),比能量較低,因而其產業化受到制約.有機電解液電壓窗口較寬(可達2?4 V),比能量較高,是當今的研究熱點.離子液體主要由二(三氟甲基磺酰)亞胺(TFSI?)、BF4?和 PF6?等陰離子與咪唑類、吡咯類及短鏈脂肪季胺鹽類等有機陽離子構成,7它具有不易揮發、熱穩定性好及電化學窗口寬等特點,15?17但是離子液體也存在諸多缺點,如明顯較低的比電容、較高的粘度和內阻.17,18
目前商品化雙電層電容器常用的電解液為1 mol·L?1四乙基四氟硼酸銨的乙腈溶液(Et4NBF4/AN)和1 mol·L?1四乙基四氟硼酸銨的丙烯碳酸酯溶液(Et4NBF4/PC).乙腈為非質子極性溶劑,粘度低但易揮發.而丙烯碳酸酯是一種強極性溶劑,熔點低,沸點高但粘度偏高.因此,采用乙腈和丙烯碳酸酯混合溶劑發揮各自的特點,將有助于提高電解液的穩定性和電壓窗口.19?21
本文采用活性炭作為電極極化活性物質,組裝成軟包裝超級電容器,首次研究了其在1 mol·L?1三乙基甲基四氟硼酸銨(MeEt3NBF4)的AN和PC混合溶劑(質量比為1:1)中的電化學性能.MeEt3NBF4的溶解度和電化學分解電壓比Et4NBF4高,此外還具有陰陽離子半徑大小相當等特點.我們分別以(1 mol·L?1MeEt3NBF4/(AN+PC)、1 mol·L?1Et4NBF4/AN和1 mol·L?1Et4NBF4/PC)為電解液制作活性炭基超級電容器,并在3 V電壓下對它們的循環伏安、電化學阻抗譜、恒流充放電、漏電流、自放電、循環壽命和庫侖效率等電化學性能進行了系統比較.
2 實驗
2.1 實驗材料
本文使用的新型有機電解液由北京化學試劑研究所提供,電導率為40 mS·cm?1,溶劑為乙腈和丙烯碳酸酯按1:1的質量比混合而成(電池級),電解質為MeEt3NBF4,記為ME;1 mol·L?1Et4NBF4/AN溶液記為 AN(溶劑純度99.9986),1 mol·L?1Et4NBF4/PC溶液記為PC(溶劑純度99.9974),電導率分別為55.21和13.35 mS·cm?1,以上兩種電解液均來自于深圳新宙邦公司.使用導電炭黑(super C45,SC)作導電劑.粘接劑是羧甲基纖維素鈉(CMC)和丁苯橡膠(SBR)按1:2質量比混合而成,使用刻蝕鋁箔(JCC,20 μm)作為集流體以減少鋁箔與活性物質之間的接觸電阻.22
2.2 超級電容器制備方法
電極片的制備方法如下,按質量比m(AC):m(SC):m(CMC):m(SBR)=80:10:2:4的比例備料,將CMC和SBR溶于去離子水中,磁力攪拌1 h得到均勻的CMC和SBR混合溶液備用.將活性炭、導電炭黑球磨30 min混勻,加入已制備好的CMC和SBR混合溶液后再球磨2 h混勻.將混勻的漿料用自動涂布機均勻地涂在鋁箔上,涂布厚度為100 μm.然后,轉移至干燥箱中,在空氣氣氛下,80°C干燥12 h.
超級電容器制備方法為:將極片沖成3.5 cm×4 cm的矩形.再用壓片機以相同的壓力將極片壓實,用膠帶將極片和隔膜按正極-隔膜-負極的順序裹好.用超聲點焊機在極耳處焊接上引出電極,置于鋁塑包內,然后100°C真空干燥24 h.快速轉移至氬氣手套箱(水分含量和氧氣含量均小于1.0×10?7)中,分別注入三種不同的電解液,移出手套箱,真空封口.
2.3 電化學測試
在上海辰華公司CHI660c電化學工作站上進行循環伏安(CV)測試.使用Autolab對其進行電化學阻抗譜(EIS)測試,交流信號振幅為10 mV,頻率范圍為0.01 Hz?100 kHz.充放電實驗在美國Arbin公司MSTAT4電化學測試系統上完成.漏電流測試方法為先以1 A·g?1(質量按正負極極化活性物質AC總質量計算,下同)的電流密度,恒流充至額定電壓,再以額定電壓恒壓充電2 h后,測得的電流作為漏電流,漏電流測試同樣在Arbin電化學測試系統上完成.循環性能測試在武漢Land電化學測試系統上進行,充放電電流密度為1A·g?1.
特制三電極體系是在兩電極體系的基礎上,在工作電極和對電極的外側各添加一片新的AC電極,然后將兩片新的AC電極通過同一引出電極引出作為參比電極,AC參比電極用隔膜與工作電極和對電極隔開.
超級電容器的電極材料比電容Cs(單位:F·g?1)、內阻R(單位:Ω)和庫侖效率η可以利用恒流充放電實驗,按以下公式求得.23

其中:I為恒流充放電電流(單位:A),M為正負極極化活性物質(AC)總質量(單位:g),Δt為放電時間(單位:s),ΔU為充放電電流換向時的電壓降(單位:V),U?為電壓窗口上限與ΔU之差(單位:V),tdis為放電時間(單位:s),tch為充電時間(單位:s).
3 結果和討論
3.1 循環伏安
循環伏安曲線可用來判斷超級電容器的內阻特性,表征超級電容器的容量特性和確定電解液的電壓窗口.對于理想的電容行為,CV曲線呈現出對稱的矩形,由于超級電容器的內阻,CV曲線的矩形形狀將發生扭曲(矩形的兩豎線變成彎曲的斜線),內阻越大,扭曲越嚴重.由于矩形的面積與充電容量和放電容量之和成正比,所以,矩形面積越大說明超級電容器的容量越高.同時當CV曲線電流出現較快增長時,表明超級電容器內電解液開始分解.
圖1(a)是ME電解液在不同電壓窗口下的CV曲線,掃描速率是20 mV·s?1,縱坐標為充放電電流(單位:A).從圖中可以看出,在2.5?3.1 V的截止電壓下,電流無明顯增加,同時CV曲線的扭曲程度未明顯加強,說明超級電容器的內阻無明顯增大并且ME電解液在3.1 V下未發生明顯分解.在3.3和3.5 V下,電流有明顯提高,同時扭曲程度明顯加強,說明電解液分解程度更加嚴重,分解產物使超級電容器的內阻開始增大.
圖1(b)是三種超級電容器在3 V下的CV曲線,掃描速率是20 mV·s?1,縱坐標為電流密度(充放電電流與極化活性物質總量之比,單位:A·g?1).從圖中可以看出,AN和ME電解液的CV曲線大致重合,這說明AN和ME電解液有非常相似的電容特性.同時,AN和ME電解液較PC的CV曲線扭曲程度更小,這說明AN和ME電解液的內阻更低.由于PC相對嚴重的扭曲,其CV曲線的面積要小于AN和ME電解液,可以得知PC下的比電容將不及AN和ME下的比電容,并且AN的CV曲線面積比ME電解液的CV曲線面積稍大,可知,AN的比電容較ME電解液稍大.
圖1(c)是ME電解液在不同掃描速率(20、50、100、200 mV·s?1)下的CV曲線,電壓窗口為0?3 V.可見,ME電解液在200 mV·s?1的掃描速率下依然能夠保持較好的矩形形狀,可知,在這一電解液下有較好的倍率特性,這歸功于這種電解液有較高的電導率(40 mS·cm?1).
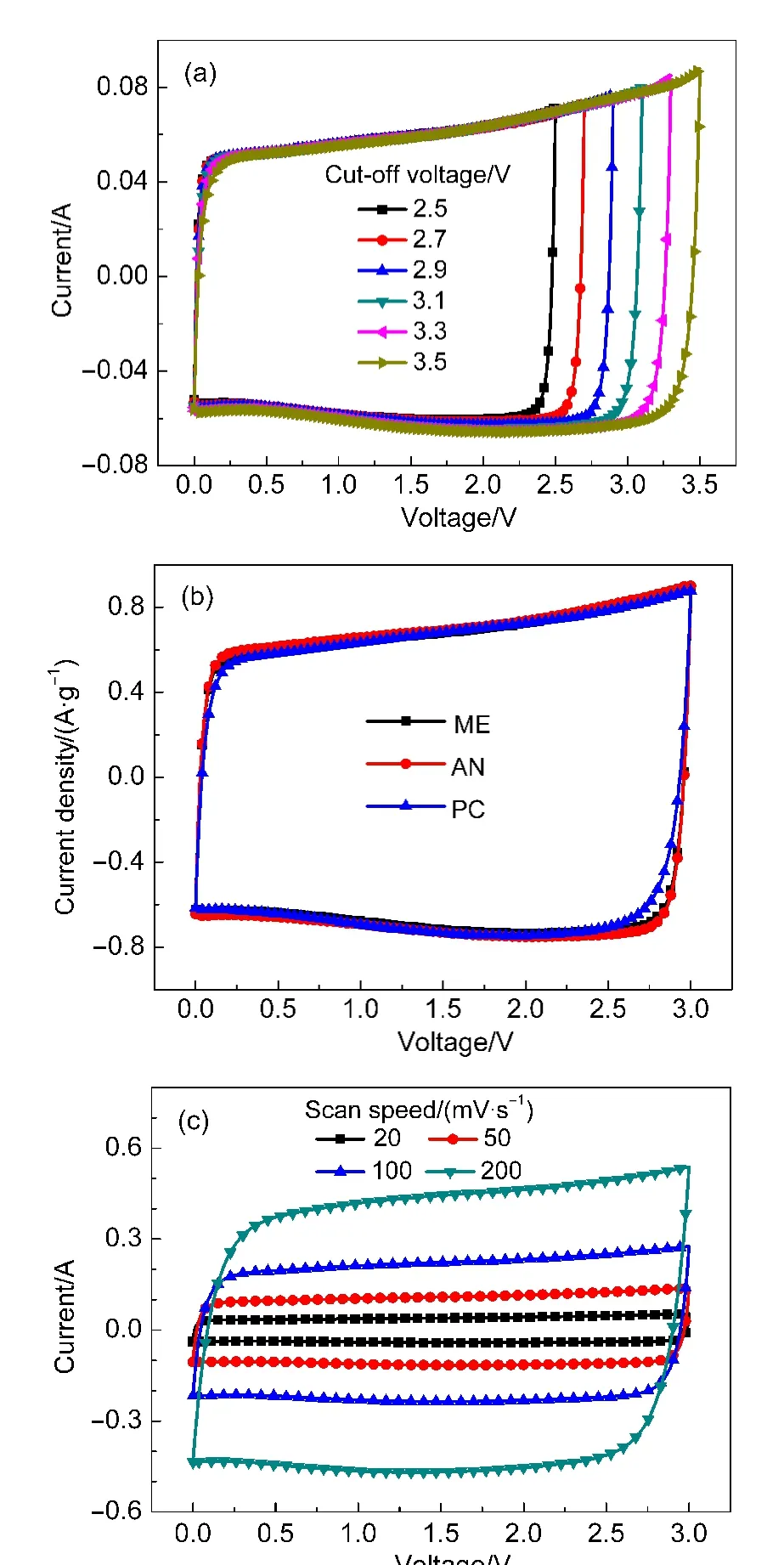
圖1 超級電容器的循環伏安(CV)曲線Fig.1 Cyclic voltammetry(CV)curves of supercapacitors
3.2 電化學阻抗譜
電化學阻抗譜(EIS)的測量方法是,通過施加某一頻率ω的正弦電壓 ?E(ω)=?Emaxejωt, 得到同頻率ω下的正弦電流 ?I(ω)=?Imaxejωt-φ,輸出信號 ?E(ω)與相應輸入信號?I(ω)的比值就是在該頻率ω下的交流阻抗:其中,″分別是Z(ω)的實部和虛部.
使用不同電解液的超級電容器的電化學阻抗譜如圖2(a)所示,振幅均為10 mV,頻率為0.01 Hz?100 kHz.由圖可見,三種超級電容器的EIS形狀相似,主要由高頻區的半圓弧、中頻區的45°斜線和低頻區的豎線三部分組成.高頻區的半圓弧與電荷轉移阻抗和極化阻抗有關.24,25中頻區的45°斜線主要由多孔活性炭電極的分布電容和分布電阻引起,低頻區的豎線由超級電容器的雙電層電容引起,豎線越接近豎直,說明電容的電容性越好.曲線與實軸的交點被稱為溶液電阻RE,其主要是電解液的內阻,另外還包括了活性炭電極的內阻、活性炭電極與集流體間的接觸電阻.半圓弧所引起的電阻增加部分被稱為電荷轉移電阻Rct.電荷轉移電阻與溶液電阻一起構成等效串聯電阻(ESR).活性炭多孔結構里的電解液電阻被稱為等效分布電阻(EDR),26低頻區豎線的延長線與實軸的交點被稱為超級電容器的總內阻(Rtotal),Rtotal=ESR+EDR.
由圖2可知,三種電容的總內阻Rtotal按PC、ME、AN的順序依次減小,同時溶液電阻RE和EDR也按PC、ME、AN的順序依次減小,這與三種電解液的電導率大小是相吻合的,ME電解液電導率居中,符合商用電解液電導率要求.PC電解液有較低的電導率原因在于其較高的粘度.27,28同時,在低頻區,三條斜線相互平行,說明三種電容的電容特性接近.
圖2(b)是ME電解液在不同截止電壓下充放電后的EIS,測試方法為:將超級電容器依次在遞增的截止電壓下充放電,每一截止電壓下充放電5周后,測試超級電容器的EIS.由圖可見,隨著截止電壓的提高,超級電容器的電荷轉移電阻Rct不斷增大,這主要是由電解液在較高截止電壓下的分解產物所引起.同時電荷轉移電阻Rct的增長速度也在增大,這說明超級電容器的電解液分解加快.例如,截止電壓從2.5 V提高到2.6 V時的Rct增量要明顯小于電壓從3 V提高到3.1 V的Rct增量.
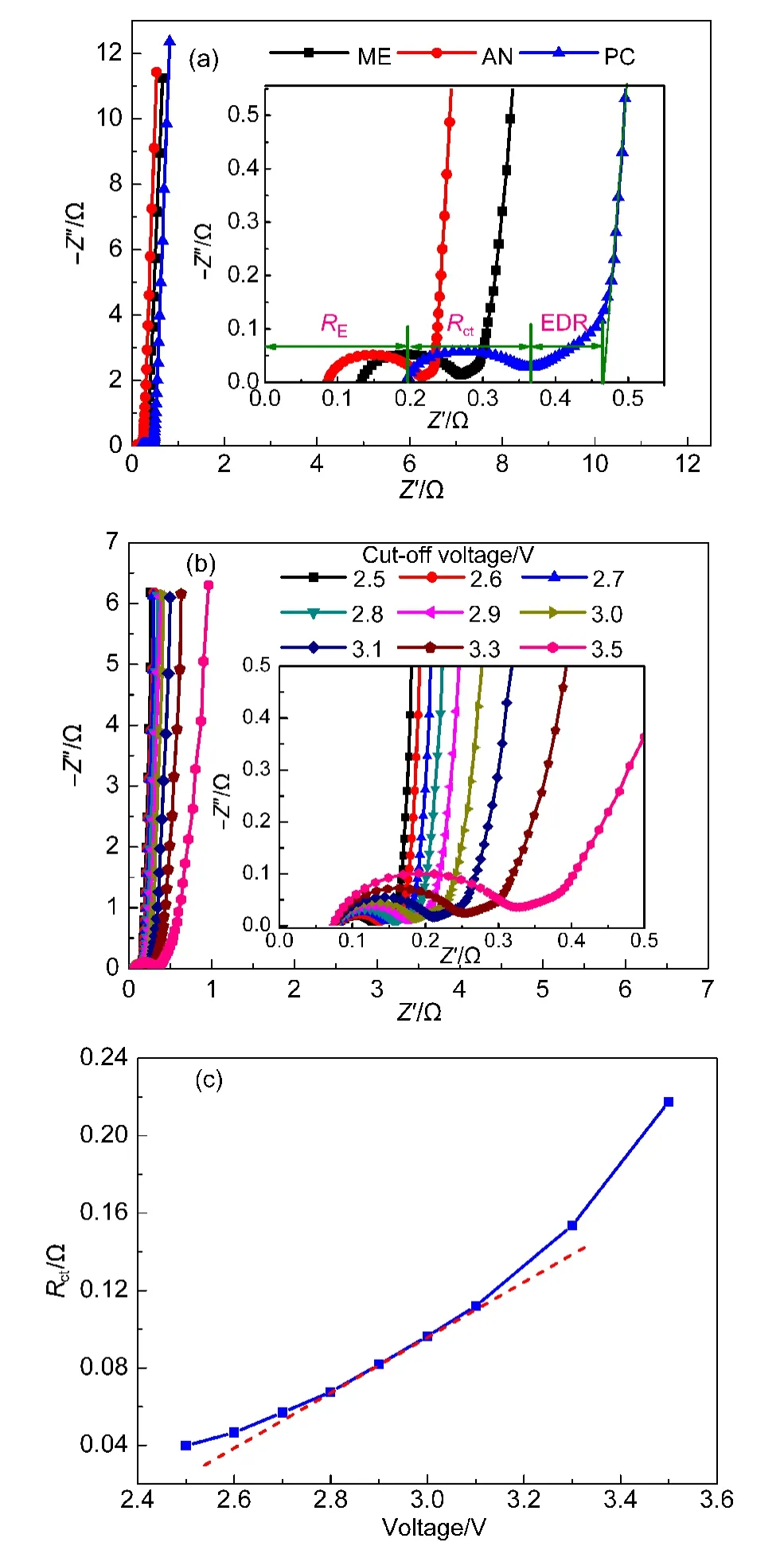
圖2 超級電容器的電化學阻抗譜(EIS)和電荷轉移電阻(Rct)Fig.2 Electrochemical impedance spectroscopy(EIS)and charge transfer resistance(Rct)of supercapacitors
為了定量分析Rct增量變化情況,用Zview軟件求出圖2(b)中Rct,圖2(c)是超級電容器在不同截止電壓下充放電后的電荷轉移電阻Rct大小.由圖可知,電荷轉移電阻Rct隨截止電壓的增大而不斷增大,同時曲線的斜率隨截止電壓的增大而不斷增大,說明隨著截止電壓的提高,電解液的分解程度越來越嚴重,尤其是在3.3和3.5 V下,電荷轉移電阻Rct增大十分明顯,這與ME電解液在不同截止電壓下的EIS和CV曲線取得很好的一致性.
3.3 恒流充放電和比電容
圖3(a)為ME電解液在不同電流密度(相對于正負極AC總質量)下的恒流充放電曲線,電流密度分別為0.2、0.5、1、2、5、10 A·g?1,電壓窗口為0?3 V.根據公式(1)-(3)可計算出三種超級電容器在不同電流密度下的比電容Cs、內阻R.經計算,等效內阻R隨電流密度的變化不大,但不同電容的內阻不同,ME電解液為0.204 Ω,AN為0.176 Ω,PC為0.301 Ω,這表明ME電解液電導率居中,這與三者的EIS和電導率是一致的.
圖3(b)是3 V下三種超級電容器的比電容Cs隨電流密度的變化關系,由圖可知,在200 mA·g?1的電流密度下,三種超級電容器ME、AN和PC的比電容分別為144.70、145.92 和142.03 F·g?1,AN的比電容最高,PC的比電容最低,這主要是因為AN的分子較PC分子小,所以AN的溶劑化離子比PC的溶劑化離子更小,更易深入活性炭多孔電極的微孔.在電流密度低于2 A·g?1時,三種超級電容器的比電容均快速下降.隨著電流密度的繼續增大,ME和AN的下降趨勢十分相似,下降程度要明顯低于PC.
圖3(c)是三種電解液下三電極體系超級電容器在3V下的充放電曲線,充放電電流密度為0.2A·g?1.可見,放電時間AN>ME>PC,即比電容AN>ME>PC,ME的比電容居中,AN的比電容比PC大,還可以看出,三種超級電容器的正極的電位窗口比負極的電位窗口窄,由此可知,三種電解液下,正極的比電容比負極的比電容大,經計算ME、AN、PC三種超級電容器的正負極比電容之比分別為:1.109、1.088、1.149,ME電解液有適中的正負極比電容比.
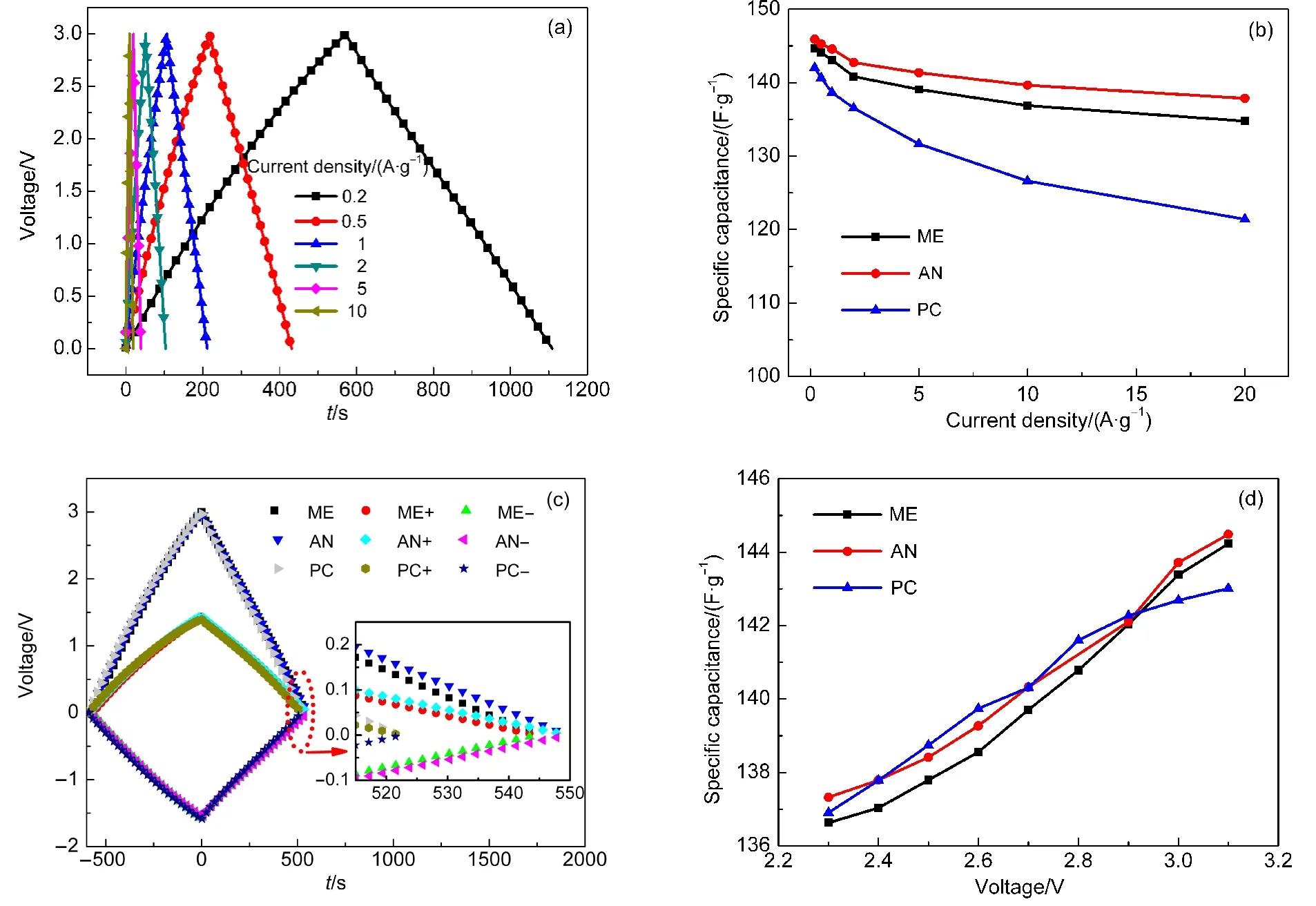
圖3 超級電容器的恒流充放電曲線(a,c)和比電容曲線(b,d)Fig.3 Galvanostatic charge-discharge curves(a,c)and specific capacitance curves(b,d)of supercapacitor
圖3(d)是不同截止電壓下,三種超級電容器的比電容變化情況,可見,隨著截止電壓的提高,三種電解液的比電容均有所提高,比電容與工作電壓大致為線性關系,這一現象可由CV曲線加以解釋:由截止電壓升高后的CV曲線可見,在升高前的截止電壓范圍內的電流要明顯低于電壓升高范圍的電流,這樣整個電壓窗口的平均值將有所增加,即比電容有所提高.比如由圖1(a)可見,3.1 V下的CV曲線較2.9 V下的CV曲線的電壓升高范圍為2.9?3.1 V,在這一范圍內,電流較升高前的截止電壓范圍內(0?2.9 V)的電流明顯要大,3.1 V截止電壓下的CV曲線的電流均值較2.9 V截止電壓下的CV曲線的均值就有所提高.同時還可見,在3 V的工作電壓下,三種電解液的比電容大小順序為AN>ME>PC,這與在3 V下CV曲線所觀測到的結論是完全一致的.
3.4 漏電流和自放電測試
漏電流主要由以下三個方面引起:(1)由電極或電解質中存在的雜質引起的微電化學電池電流;(2)由電勢梯度和離子濃度差引起的雙電層電荷擴散電流;(3)集流體或殼體毛刺刺穿隔膜所引起的微短路電流.漏電流測試的主要作用是衡量超級電容器的絕緣性能.如圖4可知,恒壓充電大約前1000 s,PC的漏電流密度要高于ME和AN,但是PC的漏電流密度下降速度比ME和AN要快.大約在第3000 s左右,三種超級電容器的漏電流密度已基本穩定.在2 h時,三種超級電容器漏電流密度依次為4.45、7.43、4.41 mA·g?1,可見ME電解液的漏電流密度與PC極為相似,AN的漏電流密度要明顯比ME和PC的高.
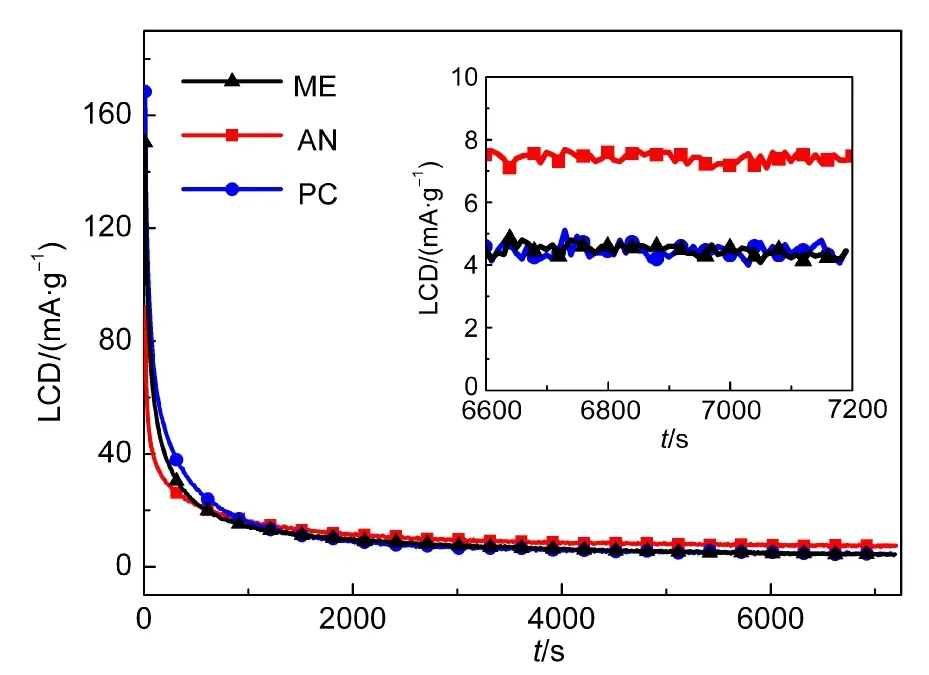
圖4 不同電解液超級電容器的漏電流密度(LCD)和放大圖(插圖)Fig.4 Leakage current density(LCD)of supercapacitors with different electrolytes and amplified plots(inset)
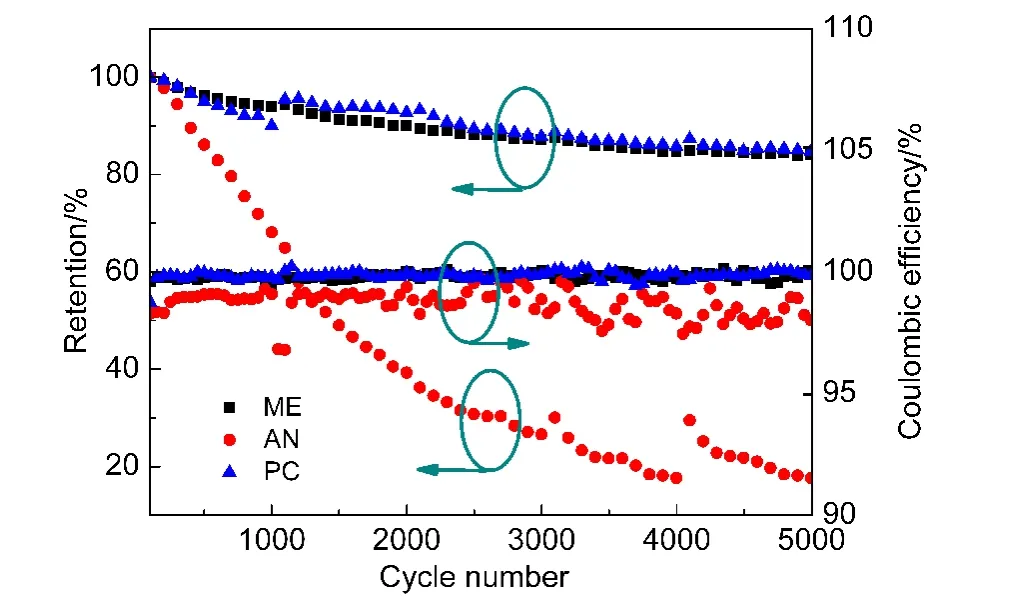
圖5 超級電容器比電容保持率和庫侖效率與循環次數的關系Fig.5 Evolution of the specific capacitance retention and coulombic efficiency versus the number of cycles
自放電測試主要目的是為了衡量超級電容器長時間存儲電荷的能力,本文自放電的測試方法是:在超級電容器漏電流測試結束后(超級電容器此時電壓為3 V),將超級電容器靜置12 h,然后測量超級電容器保持的電壓.漏電流是引起超級電容器電壓隨擱置時間降低的主要原因.三種電解液下的電壓分別為:2.2778、0.9623、2.4267 V,可見,AN電壓損失最大,這與其有較大的漏電流相吻合.ME電解液與PC相似,但比PC稍差.
3.5 循環壽命和庫侖效率
循環壽命是衡量超級電容器質量好壞的一個重要指標.隨著充放電循環的增加,電解液不斷分解,累積的電解液分解產物會堵塞活性炭發達的孔洞,導致極片的有效表面積減少,從而使電容量下降.29,30
圖5是三種超級電容器在3 V下的循環壽命和庫侖效率曲線,其充放電電流密度為1 A·g?1.超級電容器的容量隨著循環次數的增加而減小,尤其是在開始1000周衰減較大,AN在3 V下的循環表現最差,PC稍微優于ME但總體差別不大.在第5000周處,ME電解液容量保持率為83.90%,PC保持率為84.63%,AN保持率為17.39%,可見AN的循環壽命遠不及ME和PC.同時,由庫侖曲線知,AN的效率約為98.5%,比ME和PC明顯要差,ME和PC的效率約為99.8%,可見ME的庫侖效率與PC相似.
4 結 論
使用一種新型有機混合電解液(三乙基甲基四氟硼酸銨/(丙烯碳酸酯+乙腈))和兩種傳統的傳統的電解液(四乙基四氟硼酸銨/丙烯碳酸酯和四乙基四氟硼酸銨/乙腈),分別制作成活性炭基軟包裝超級電容器,并在3 V下對三種超級電容器進行了綜合比較,經比較發現,新型的混合電解液明顯吸收了AN和PC各自的優點,電導率高達40 mS·cm?1,且比電容大、漏電流小、循環壽命好、庫侖效率高,綜合性能優異.
致謝: 感謝北京化學試劑研究所提供本文所使用的新型有機電解液.
(1)Yu,P.;Zhang,X.;Chen,Y.;Ma,Y.;Qi,Z.Mater.Chem.Phys.2009,118,303.doi:10.1016/j.matchemphys.2009.07.057
(2) Zhang,D.;Zhang,X.;Chen,Y.;Wang,C.;Ma,Y.;Dong,H.;Jiang,L.;Meng,Q.;Hu,W.Phys.Chem.Chem.Phys.2012,14,10899.doi:10.1039/c2cp41051f
(3)Ammam,M.;Fransaer,J.J.Electrochem.Soc.2011,158,A14.
(4) Pandolfo,A.G.;Hollenkamp,A.F.J.Power Sources 2006,157,11.doi:10.1016/j.jpowsour.2006.02.065
(5) Sun,X.;Zhang,X.;Zhang,H.;Zhang,D.;Ma,Y.J.Solid State Electrochem.2012,16,2597.doi:10.1007/s10008-012-1678-7
(6) Kurig,H.;Janes,A.;Lust,E.J.Electrochem.Soc.2010,157,A272.
(7) Zhong,H.X.;Zhao,C.B.;Luo,H.;Zhang,L.Z.Acta Phys.-Chim.Sin.2012,28,2641.[仲皓想,趙春寶,駱 浩,張靈志.物理化學學報,2012,28,2641.]doi:10.3866/PKU.WHXB201207181
(8) Lu,W.;Hartman,R.;Qu,L.T.;Dai,L.M.J.Phys.Chem.Lett.2011,2,655.doi:10.1021/jz200104n
(9) Kim,B.;Chung,H.;Kim,W.Nanotechnology 2012,23.
(10) Sun,X.Z.;Zhang,X.;Zhang,D.C.;Ma,Y.W.Acta Phys.-Chim.Sin.2012,28,367.[孫現眾,張 熊,張大成,馬衍偉.物理化學學報,2012,28,367.]doi:10.3866/PKU.WHXB201112131
(11) Liu,P.;Verbrugge,M.;Soukiazian,S.J.Power Sources 2006,156,712.doi:10.1016/j.jpowsour.2005.05.055
(12) Portet,C.;Taberna,P.L.;Simon,P.;Flahaut,E.J.Power Sources 2005,139,371.doi:10.1016/j.jpowsour.2004.07.015
(13) Sillars,F.B.;Fletcher,S.I.;Mirzaeian,M.;Hall,P.J.Phys.Chem.Chem.Phys.2012,14,6094.doi:10.1039/c2cp40089h
(14) Lewandowski,A.;Olejniczak,A.;Galinski,M.;Stepniak,I.J.Power Sources 2010,195,5814.doi:10.1016/j.jpowsour.2010.03.082
(15) Pandey,G.P.;Kumar,Y.;Hashmi,S.A.Indian J.Chem.Sect.A-Inorg.Bio-Inorg.Phys.Theor.Anal.Chem.2010,49,743.
(16) Kurig,H.;Vestli,M.;Tonurist,K.;Janes,A.;Lust,E.J.Electrochem.Soc.2012,159,A944.
(17) Ruiz,V.;Huynh,T.;Sivakkumar,S.R.;Pandolfo,A.G.RSC Adv.2012,2,5591.doi:10.1039/c2ra20177a
(18) Balducci,A.;Dugas,R.;Taberna,P.L.;Simon,P.;Plee,D.;Mastragostino,M.;Passerini,S.J.Power Sources 2007,165,922.doi:10.1016/j.jpowsour.2006.12.048
(19)Tyunina,E.Y.;Afanasiev,V.N.;Chekunova,M.D.Journal of Chemical&Engineering Data 2011,56,3222.doi:10.1021/je200309v
(20) Moumouzlas,G.;Panopoulos,D.K.;Ritzoulis,G.Journal of Chemical and Engineering Data 1991,36,20.doi:10.1021/je00001a006
(21) Borenstien,A.;Noked,M.;Okashy,S.;Aurbach,D.J.Electrochem.Soc.2013,160,A1282.
(22)Wang,G.X.;Shao,Z.P.;Yu,Z.L.Nanotechnology 2007,18,205705.
(23)Chen,H.;Wang,F.;Tong,S.S.;Guo,S.L.;Pan,X.M.Appl.Surf.Sci.2012,258,6097.doi:10.1016/j.apsusc.2012.03.009
(24)Liu,X.M.;Zhang,R.;Zhan,L.;Long,D.H.;Qiao,W.M.;Yang,J.H.;Ling,L.C.New Carbon Mater.2007,22,153.doi:10.1016/S1872-5805(07)60015-8
(25) Ferg,E.;Rossouw,C.;Loyson,P.J.Power Sources 2013,226,299.doi:10.1016/j.jpowsour.2012.10.087
(26) Kotz,R.;Carlen,M.Electrochim.Acta 2000,45,2483.doi:10.1016/S0013-4686(00)00354-6
(27) Lust,E.;J?nes,A.;Arulepp,M.J.Electroanal.Chem.2004,562,33.doi:10.1016/j.jelechem.2003.07.034
(28) Arulepp,M.;Permann,L.;Leis,J.;Perkson,A.;Rumma,K.;J?nes,A.;Lust,E.J.Power Sources 2004,133,320.doi:10.1016/j.jpowsour.2004.03.026
(29) Ruch,P.W.;Cericola,D.;Foelske-Schmitz,A.;K?tz,R.;Wokaun,A.Electrochim.Acta 2010,55,4412.doi:10.1016/j.electacta.2010.02.064
(30) Ruch,P.W.;Cericola,D.;Foelske,A.;K?tz,R.;Wokaun,A.Electrochim.Acta 2010,55,2352.doi:10.1016/j.electacta.2009.11.098
