蘇丹紅Ⅰ磁性分子印跡聚合物的制備及其分離分析應用
2013-10-08 00:51:52曾延波張祖磊陳智棟
分析測試學報 2013年6期
馬 潔,黃 靜,曾延波,張祖磊,張 劍,陳智棟,李 蕾*
(1.常州大學 化學化工學院,江蘇 常州 213016;2.嘉興學院 生物與化學工程學院,浙江 嘉興 314001)
分子印跡聚合物具有預定選擇性、化學性質穩定、對環境的耐受性好和制備簡單等優點,因此在固相萃取、傳感器、催化等領域得到廣泛應用[1-4]。磁性分離是在磁場的作用下依據物質的磁性差異,將磁性組分與非磁性組分分離的一種技術[5]。將磁性分離技術與分子印跡技術相結合制備的磁性分子印跡聚合物(M-MIPs),兼具分子印跡聚合物和磁性材料的特點,可選擇性分離富集分析物,使之在外磁場的作用下快速與基質分離。近年來基于磁性材料的分子印跡聚合物逐漸成為研究熱點。
蘇丹紅是一種人工合成的工業染料,國際癌癥機構將其列為三類致癌物質[6],但一些不法食品企業將其作為食品色素,嚴重危害消費者健康。目前食品中蘇丹紅Ⅰ的檢測方法主要有HPLC法、GCMS聯用法、電化學檢測法等[7-10],而這些方法大都需要繁瑣的樣品前處理過程。用分子印跡聚合物作為萃取材料,可簡化樣品前處理過程[11-14],但通常需將其離心分離或者用于填充柱,分離過程仍較繁瑣,而M-MIPs在外磁場作用下可與基質快速分離,因此,M-MIPs的使用降低了樣品預處理的復雜性,提高了方法的可靠性。本文以3-(異丁烯酰氧)丙基三甲氧基硅烷(MPS)修飾的 Fe3O4為磁性組分,以蘇丹紅Ⅰ為模板分子,甲基丙烯酸為功能單體,乙二醇二甲基丙烯酸酯為交聯劑,制備了磁性分子印跡聚合物,并將其用于辣椒粉提取液中蘇丹紅Ⅰ的分離富集,在外磁場下實現快速分離,用甲醇洗脫,并結合液相色譜法進行檢測。該方法具有操作簡單、選擇好、回收率高等優點。
1 實驗部分
1.1 儀器與試劑
UV-2550型紫外可見分光光譜儀(日本島津公司),Agilent 1200型高效液相色譜儀(美國安捷倫科技有限公司),NEXUS470型傅立葉變換紅外光譜儀(美國熱電尼高力公司),Hitachi s-4800型掃描電子顯微鏡(日本日立公司),JEM-200CX型透射電子顯微鏡(日本JEOL公司),Lake Shore 7410型振動樣品磁強計(美國Lake Shore Cryotronics公司)。
FeCl3·6H2O、FeSO4·7H2O、3-(異丁烯酰氧)丙基三甲氧基硅烷(MPS)(分析純,上海晶純試劑有限公司);蘇丹紅Ⅰ、蘇丹紅Ⅱ、蘇丹紅Ⅲ、氨水、乙酸(分析純)及甲基丙烯酸(MAA,化學純,使用前提純)均購于國藥集團化學試劑有限公司;乙二醇二甲基丙烯酸酯(EGDMA,98%)、偶氮二異丁腈(AIBN,98%)(使用前提純,上海百靈威有限公司);甲醇、氯仿、乙腈(分析純);超純水(實驗室自制);辣椒粉購于當地超市。
1.2 蘇丹紅Ⅰ磁性分子聚合物(M-MIPs)的制備
取2 g制備的Fe3O4顆粒[15]于三口燒瓶中,加入38 mL無水乙醇和2 mL水,再加入0.4 mL MPS,超聲0.5 h后,在回流溫度下攪拌反應3 h[16]得到MPS修飾的Fe3O4顆粒。
取1 mmol(0.25 g)蘇丹紅Ⅰ溶于50 mL乙腈中,加入4 mmol(0.34 mL)MAA超聲0.5 h后,加入10 mmol(1.89 mL)交聯劑EGDMA、1 g MPS修飾的Fe3O4和0.1 g引發劑AIBN,N2氛圍下65℃聚合反應24 h。空白印跡聚合物(M-NMIPs)的制備方法同上,只是制備過程中未加模板分子。
M-MIPs用索氏提取器抽提(抽提液為體積比為9∶1的甲醇-乙酸)直至檢測不到模板分子,用甲醇洗去殘留乙酸后,于60℃真空干燥過夜。
1.3 聚合物的吸附性能測試
1.3.1 靜態吸附性能研究 稱取20 mg M-MIPs加入到錐形瓶中,然后分別加入5 mL不同濃度(0.3~3.0 mmol/L)的蘇丹紅Ⅰ-乙腈標準溶液,室溫振蕩12 h后,用磁鐵吸附M-MIPs,取清液用紫外可見分光光度法測定平衡時蘇丹紅Ⅰ的濃度,根據吸附前后的濃度變化,計算M-MIPs對不同濃度蘇丹紅Ⅰ溶液的吸附量Q,其公式為Q=(C0-Ce)V/m,其中Q為靜態平衡時的吸附量(μmol/g),C0為底物的初始濃度(mmol/L),Ce為吸附平衡時清液的濃度(mmol/L),V為底物溶液的體積(L),m為磁印跡材料的加入量(g)。M-NMIPs使用上述方法測定結合量并與M-MIPs相比較。
1.3.2 選擇性吸附實驗 為研究聚合物的選擇性,將蘇丹紅Ⅱ和蘇丹紅Ⅲ(結構式見圖1)作為底物,用平衡吸附實驗研究聚合物的識別性能。將20 mg M-MIPs放入5 mL分別含有1.0 mmol/L的蘇丹紅Ⅰ、蘇丹紅Ⅱ和蘇丹紅Ⅲ的乙腈標準溶液中,室溫振蕩12 h后,用磁鐵吸附聚合物,取清液用紫外可見分光光度法測定平衡時底物的濃度,根據吸附前后的濃度變化,計算聚合物對3種底物的結合量,M-NMIPs使用同樣方法測定。
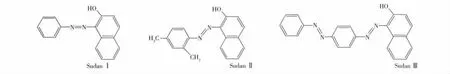
圖1 蘇丹紅Ⅰ、蘇丹紅Ⅱ和蘇丹紅Ⅲ的分子結構式Fig.1 Molecular structures of SudanⅠ,SudanⅡand SudanⅢ
1.4 樣品加標處理及萃取過程
在1 g辣椒粉表面噴灑1 mL的蘇丹紅Ⅰ-乙腈溶液(加標濃度為1.5~60 μg/g),靜置過夜后,用5 mL乙腈超聲提取10 min,離心取上清液,殘渣再重復提取2次,合并提取液。
將60 mg M-MIPs加入到提取液中,振蕩吸附12 h后用強磁鐵將吸附劑與清液分離。用2 mL甲醇溶液對吸附劑超聲洗脫,洗脫液經0.45 μm有機濾膜過濾后,再使用高效液相色譜進行檢測,計算回收率。
色譜條件:Agilent 1200 HPLC系統包括G1322A脫氣機,G1311A四元泵,G1316A柱保溫箱,G1315D二極管陣列檢測器,G1329A進樣器。色譜柱:反相Extend-C18柱(5 μm,250 mm×4.6 mm),柱溫:35℃;流動相:50%甲醇水溶液-色譜純甲醇(10∶90);檢測波長:478 nm;流速:1.0 mL/min;進樣體積:10 μL。
2 結果與討論
2.1 磁性分子印跡聚合物的制備
本文利用MPS修飾的Fe3O4粒子為磁性組分,制備了蘇丹紅Ⅰ的磁性分子印跡聚合物,具體制備路線及應用如圖2所示。
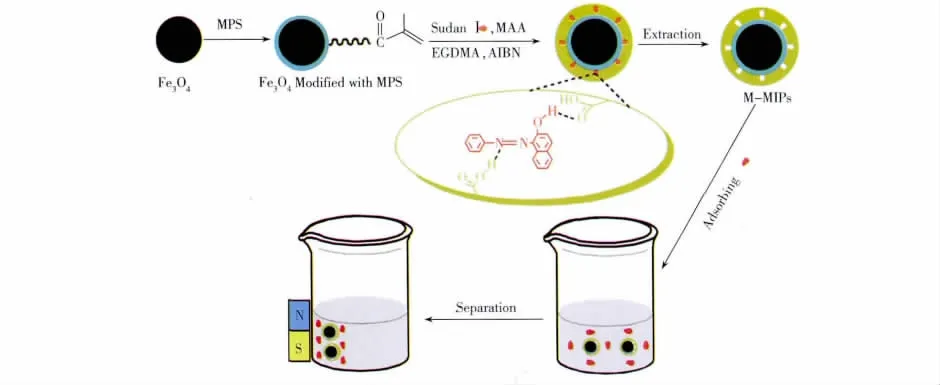
圖2 蘇丹紅ⅠM-MIPs的制備與應用過程Fig.2 Schematic diagram of the fabrication and application process of SudanⅠM-MIPs
2.2 紅外光譜分析
圖3是各‖種聚合物的紅外表征圖譜。2 919 cm-1和2‖ 857 cm-1是飽和C—H的伸縮振動峰,1 750 cm-1是MPS中CO的伸縮振動峰,1 620 cm-1為MPS中CC雙鍵的伸縮振動峰,1 123 cm-1為MPS中Si—O鍵的伸縮振動峰,580 cm-1處為Fe3O4中Fe—O鍵的特征吸收峰。對比曲線a、b可知MPS已經成功結合到Fe‖3O4粒子的表面[16-17]。M-MIPs曲線(c)中,1 460、1 390 cm-1處是甲基的面內彎曲振動峰,聚合物中CO的振動峰(1 732 cm-1)顯著增強。
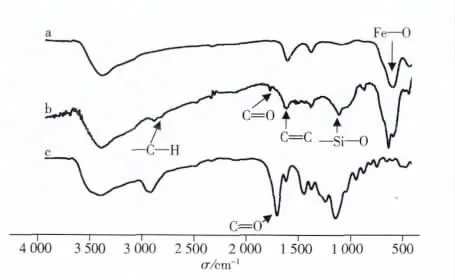
圖3 Fe3O4納米顆粒(a)、MPS修飾的Fe3O4(b)和M-MIPs(c)的紅外光譜圖Fig.3 FT-IR spectra of Fe3O4nanoparticles(a),Fe3O4modified with MPS(b)and M-MIPs(c)
2.3 磁性分子印跡聚合物的形貌結構分析
用掃描電鏡/透射電鏡(SEM/TEM)對各磁性微球的形貌結構進行分析。由圖4A中Fe3O4的SEM/TEM圖可以看到,用共沉淀法制備的Fe3O4磁性顆粒呈規則的球形結構,且粒徑比較均勻。由圖4B可以看到,由于在MPS修飾的Fe3O4微球表面進行印跡聚合作用,使得部分M-MIPs微球稍有粘連現象;從M-MIPs的TEM圖可以看到,其外殼薄膜厚度較大,這是由于聚合反應過程中參與反應的有機物的量很多,從而導致聚合物的有機物層變厚[18],說明已成功制備得到M-MIPs。
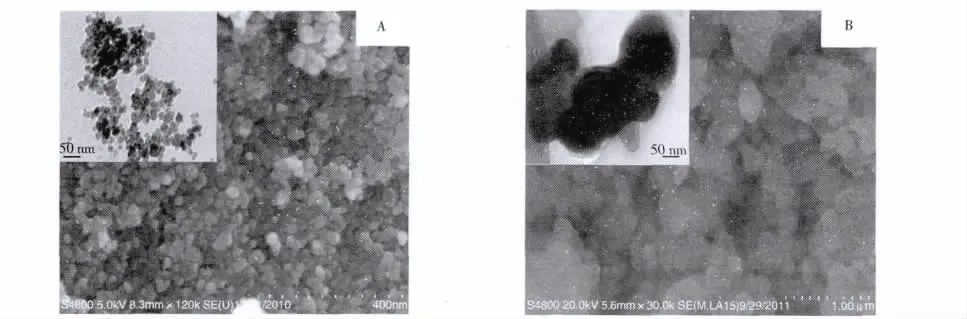
圖4Fe3O4納米顆粒(A)及M-MIPs(B)的TEM/SEM圖Fig.4 TEM/SEM images of Fe3O4nanoparticles(A)and M-MIPs(B)
2.4 磁性分子印跡聚合物的磁性能表征
考察了幾種材料的常溫磁滯回歸曲線(見圖5A)。由圖可知,由于Fe3O4顆粒表面包裹物的增加,Fe3O4顆粒、MPS修飾的Fe3O4、M-MIPs的飽和磁化強度依次降低。圖5B中左側為MMIPs的分散液,右側為在外磁場下M-MIPs的分散液,可以看出M-MIPs具有良好的磁性能,在強磁鐵的作用下,分散了M-MIPs的溶液立刻澄清,表明M-MIPs可滿足磁分離需求。
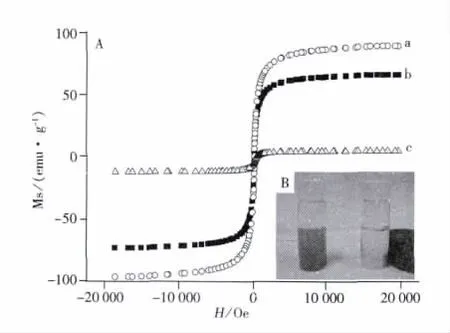
圖5 幾種材料的常溫磁滯回歸曲線(A)及M-MIPs在外磁場下的分離示意圖(B)Fig.5 The magnetization curves of several materials(A)and the separation process of M-MIPs in the presence of an external magnetic field(B)a.Fe3O4nanoparticles,b.Fe3O4modified with MPS,c.M-MIPs
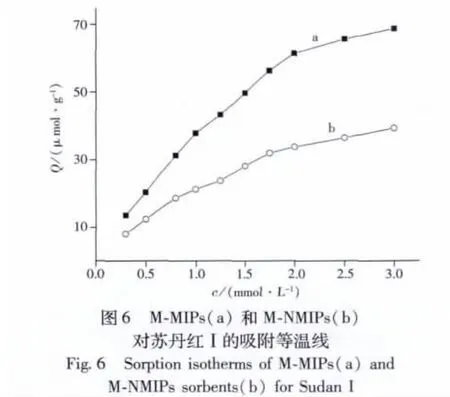
2.5 吸附等溫線
采用靜態平衡法,測定了印跡聚合物和非印跡聚合物對不同濃度蘇丹紅Ⅰ的吸附等溫線(見圖6)。
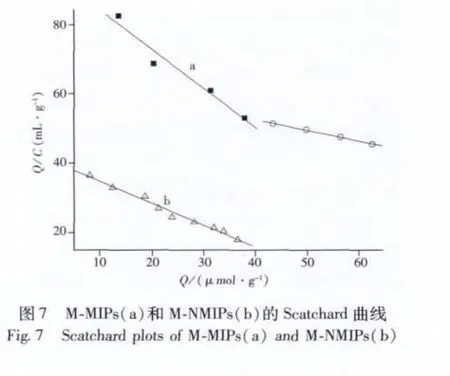
分子印跡聚合物的結合特性常用Scatchard模型[19]來評價:Q/Ce=(Qmax-Q)/Kd,式中Kd是平衡離解常數,Qmax和Q分別為M-MIPs對蘇丹紅Ⅰ的最大吸附量和理論平衡吸附量(μmol/g)。
以Q/Ce對Q作圖即得 Scatchard曲線(圖7),M-MIPs對蘇丹紅Ⅰ的吸附作用并非完全等價,而是存在兩類不同的結合位點。將圖中線性較好的兩個部分分別進行擬合,得到擬合線性方程:Q/Ce=-1.127Q+95.33(r=0.977 8)和Q/Ce=-0.308Q+64.83(r=0.998 8)。由線性方程的斜率和截距可以求得Kd1=0.89 mmol/L,Kd2=3.25 mmol/L,Qmax1=84.59 μmol/g,Qmax2=210.49 μmol/g;M-NMIPs的 Scatchard 曲線則是一條線性相關曲線,表明M-NMIPs的結合位點各向同性,所以只能形成一種非選擇性的結合位點。
2.6 選擇性吸附
采用靜態平衡法研究了聚合物對蘇丹紅Ⅰ、Ⅱ、Ⅲ的選擇吸附性,并根據公式:KD=Qe/Ce計算出對應的分配系數KD及相對選擇系數k(見表1)。式中KD為分配系數(L/g),Qe為平衡吸附量(μmol/g),Ce為吸附液中結構類似物的平衡濃度;k=KD(template)/KD(analogue)[20]。
由表1可以看出M-MIPs對蘇丹紅Ⅰ的KD最大且明顯高于M-MIPs對其他兩種物質的KD,表明其對蘇丹紅Ⅰ的吸附能力最強,而M-NMIPs對3種結構類似物的KD較小且相差不大,表明其對3種物質的吸附能力較低且相差不大;M-MIPs對蘇丹紅Ⅰ/蘇丹紅Ⅱ、蘇丹紅Ⅰ/蘇丹紅Ⅲ的k分別為2.47、2.24,明顯高于M-NMIPs對應的k,表明M-MIPs對蘇丹紅Ⅰ選擇性較好。這是因為在M-MIPs中形成了與蘇丹紅Ⅰ空間結構和結合位點相匹配的空穴,而M-NMIPs由于其功能團的分布是無序的,未能形成與蘇丹紅Ⅰ互補的識別位點,對蘇丹紅Ⅰ不具特異選擇性。
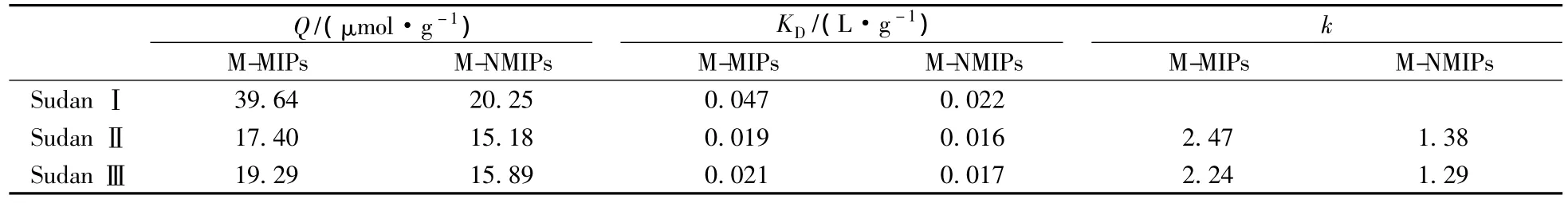
表1 聚合物的KD及k值Table 1 KDand kvalues of the polymers
2.7 方法的標準曲線與檢出限
按照“1.4”的色譜條件進行測定,以峰面積A對濃度C進行線性回歸分析。在加標濃度1.5~40 μg/g范圍內,蘇丹紅Ⅰ峰面積 (A)與濃度(C)呈良好的線性關系,其線性回歸方程為A=3.533C-6.024,相關系數(r)為0.989,方法的檢出限(S/N=3)為0.50 μg/g。
2.8 辣椒粉提取液中蘇丹紅Ⅰ的測定
按照“1.4”方法用蘇丹紅Ⅰ M-MIPs和M-NMIPs分別對加標辣椒樣品(加標濃度分別為2.4、4.8、19.2 μg/g)進行萃取處理,HPLC法測定。由表2可知,M-MIPs對應的回收率為78%~103%,相對標準偏差為2.8%~5.8%,其回收率明顯高于M-NMIPs的回收率(8%~27%),表明M-MIPs對蘇丹紅Ⅰ有特異識別性。

表2 實際樣品中蘇丹紅Ⅰ的測定(n=3)Table 2 The detection of SudanⅠin paprika sample(n=3)
用 M-MIPs和 M-NMIPs對添加19.2 μg/g和未添加蘇丹紅Ⅰ的辣椒粉進行萃取,以富集后液相峰面積與富集前液相峰面積之比計算得M-MIPs的富集倍數約為6,圖8為樣品的HPLC色譜圖。由圖可以看出,實際樣品中無蘇丹紅Ⅰ。

圖8 辣椒粉樣品(a)及加標辣椒粉樣品(b、c)經吸附萃取后洗脫液的HPLC色譜圖Fig.8 HPLC chromatograms for paprika samples(a)and spiked paprika samples(b,c)after extraction a,b:extracted with M-MIPs,c:extracted with M-NMIPs;spiked concentration of Sudan Ⅰ:19.2 μg/g
3 結論
本文以MPS修飾的Fe3O4為磁性組分,蘇丹紅Ⅰ為模板分子,甲基丙烯酸為功能單體,乙二醇二甲基丙烯酸酯為交聯劑,制備了磁性分子印跡聚合物。結合實驗表明該磁性印跡聚合物對蘇丹紅Ⅰ有較高的選擇性和吸附性。將該磁性印跡聚合物用于辣椒粉提取液中蘇丹紅Ⅰ的分離富集,用磁鐵將其與溶液快速分離,并經高效液相色譜測得樣品的回收率為78%~103%,相對標準偏差為2.8%~5.8%。
[1] Carolina Q M,Claude B,Ana M G C.Food Chem.,2012,135(2):775-779.
[2] Zhang H B,Zhang Z H,Hu Y F,Yang X,Nie L H.Chem.J.Chin.Univ.(張華斌,張朝暉,胡宇芳,楊瀟,聶利華.高等學校化學學報),2010,31(11):2141-2147.
[3] Wu B,Wang Z H,Zhao D X,Lu X Q.Talanta,2012,101:374-381.
[4] Abbate V,Bassindale A R,Brandstadt K F,Taylor P G.J.Catal.,2011,284(1):68 -76.
[5] Zeng H,Sun S H.Adv.Funct.Mater.,2008,18(3):391 -400.
[6] Stiborova M,Martinek V,Rydlova H,Hodek P,Frei E.Cancer Res.,2002,62(20):5678-5684.
[7] He R,Liao L C,Yan Y Y,Yang L,Chen L L,Jin S J,Li W J,Huang L Y.J.Instrum.Anal.(何榮,廖林川,顏有儀,楊林,陳禮莉,金圣杰,李雯佳,黃璐瑤.分析測試學報),2008,27(7):751-754.
[8] He L M,Su Y J,Fang B H,Shen X G,Zeng Z L,Liu Y H.Anal.Chim.Acta,2007,594(1):139-146.
[9] Xie X Y,Luo H Q,Li N B.Chin.J.Anal.Chem.(謝學英,羅紅群,李念兵.分析化學),2009,37:E058.
[10] Luo S X,Dai X R,Wu Y H,Lu H Y.J.Instrum.Anal.(羅宿星,代小容,伍遠輝,盧恒一.分析測試學報),2012,31(12):1562-1566.
[11] Qiao F X,Geng Y R,He C Q,Wu Y P,Pan P Y.J.Chromatogr.B,2011,879(27):2891-2896.
[12] Yan H Y,Qiao G D,Pei Y N,Long T,Ding W,Xie K.Food Chem.,2012,132(1):649-654.
[13] Xin J H,Zhao D Y,Zhang L M,Xu Z X,Zhou J,Qu D J.Chromatographia,2011,73(3/4):235 -242.
[14] Zhang Z H,Liu L,Zhou H Y,Nie L H.J.Instrum.Anal.(張朝暉,劉麗,周慧圓,聶麗華.分析測試學報),2010,29(2):136-141.
[15] Abareshi M,Goharshadi E K,Zebarjad S M,Fadafan H K,Youssefi A.J.Magn.Magn.Mater.,2010,322(24):3895-3901.
[16] Gai Q Q,Qu F,LiuZ J,Dai R J,Zhang Y K.J.Chromatogr.A,2010,1217(31):5035 -5042.
[17] Cui S,Shen X D,Lin B L.Rare Metals,2006,25:426 -430.
[18] Ji Y S,Yin J J,Xu Z G,Zhao C D,Huang H Y,Zhang H X,Wang C M.Anal.Bioanal.Chem.,2009,395(4):1125-1133.
[19] Zhou J,He X W.Anal.Chim.Acta,1999,381(1):85.
[20] Han D M,Fang G Z,Yan X P.J.Chromatogr.A,2005,1100(2):131-136.
