Au/Fe2O3-MOx催化劑在氣體純化中的應用研究
2013-10-17 06:57:26張鳳利
低溫與特氣 2013年4期
張鳳利
(1.福州市產品質量檢驗所,福州 350025;2.國家化學工業氣體產品質量監督檢驗中心,福州 350008)
當今,信息化水平已經成為衡量一個國家綜合國力的重要標志。作為信息化支柱的電子工業已發展成為當今世界的戰略性工業。而電子元器件的制造基礎是超純、高純電子化學氣體,半導體器件性能的好壞,在很大程度上取決于所用電子氣體的質量,電子氣體純度每提高一個數量級,都會極大地推動半導體器件質的飛躍[1]。在集成電路的刻蝕和清洗過程中,電子氣體中百萬分之幾的微量雜質氣體進入工序就能導致質量下降,使每個元件的信息量減少,從而使高密度集成電路產品的不合格率增加[1-3]。因此在制備高純度的氣體過程中,純化工藝顯得尤為重要。
高純永久性氣體主要包括高純 H2、O2、N2、Ar、He,主要是由工業級氣體經純化獲得。H2可以采用鈀管擴散、低溫凈化的方法獲取,但鈀管擴散不能在高壓下充瓶;He、Ar可以通過Zr-Al或Zr-V-Fe合金吸氣的工藝提純,過高的提純溫度,在高壓下進行凈化充裝,從安全方面講應該充分考慮反應器的結構和強度的計算。催化劑在制備高純氣體中發揮了重要作用。工業級永久性氣體中主要含有微量H2、O2、N2、CH4、CO 和 CO2等雜質,其純化而獲得高純氣主要是使用催化劑,將CO和H2氧化成CO2和H2O(2CO+O2=2CO2,2H2+O2=2H2O),然后再用分子篩吸附。氮氣和氬氣的純化使用氧凈化劑和水汽吸收劑。傳統的氧凈化劑使用還原銅催化劑;水汽和二氧化碳通常是用活性分子篩來捕獲。在某些情況下,使用氧化銅催化劑將CO和H2氧化成CO2和H2O,然后再用分子篩吸附。氫氣的純化與氮氣類似,均使用催化劑。典型的是用鉑催化劑將O2氧化為H2O,然后用分子篩凈化除去[4]。由此可見,氧化反應是制備高純氣體純化過程中的重要環節,氧化反應催化劑性能的好壞直接決定了氣體中雜質的脫除效率,因此獲得高性能氧化反應催化劑成為氣體純化工藝中研究的重點。
當前氧化反應催化劑研究主要鎖定在負載型金屬催化劑的研究。Pt、Ru、Pd、Co、Cu、Ag、Fe、Mo 和Au等金屬或金屬的氧化物對CO吸附能為20~50 kcal/mol,具有中等程度的吸附能力,能夠促進CO的吸附和氧化,成為活性組分研究考察的對象;Fe2O3、La2O3、V2O3、CeO2、ZrO2等具有氧化還原的性質,可產生介穩態吸附氧原子,氧化被吸附的CO并放出氫。其中Pt系(PGM)和Au等負載型貴金屬催化劑是近10年來的研究熱點,但研究發現:PGM/CeO2體系催化劑在富氫氣氛下,由于Pt系金屬與載體CeO2間存在強相互作用,使得載體CeO2被不可逆還原,催化劑在250℃下就出現線性劣化失活[5],這個問題同樣制約了以Pt系金屬為活性組分的其它負載型催化劑在純化器中的應用。而負載型Au催化劑中Au與載體間的相互作用較為適中,且Au資源較為豐富,在價格上具有一定的優勢,相對于Pt系催化劑更有研究前景[6]。基于Fe2O3本身具有氧化還原性能,而CeO2具有獨特的快速價態調變性質,常作為電子助劑、結構助劑和共催化劑應用于許多催化劑體系[7]。我們的前期工作主要圍繞著以Au為活性組分,開展Au/Fe2O3、Au/CeO2等系列催化劑的CO氧化性能研究。Au/Fe2O3催化劑在0~100℃溫區具有較高的CO轉化率,但選擇性較低;通過適宜的制備方法可以制得CO選擇性氧化性能與金鐵催化劑相當的Au/CeO2-ZrO2催化劑;通過對Au/ZSM和Au/ZSM-Fe2O3催化劑的性能對比表明,催化劑的活性是Au與Fe2O3相互作用的結果;對Fe2O3/ZSM和純Fe2O3催化劑的穩定性考察表明Fe2O3也會發生失活,這些暗示Au/Fe2O3催化劑的失活可能不僅跟Au有關,跟載體發生的某種變化也有關。通過我們的初步研究,認為該類催化劑低溫活性和穩定性的提高還具有一定的潛力,有望通過進一步的改性研究,使之最終應用于制備高純氣體純化器中。
盡管對這類催化劑的失活機制有一定的共識,如Au/Fe2O3、Au/CeO2這兩種負載型Au催化劑的失活主要是由于積炭引起。然而,如何提高催化劑的穩定性,使之滿足制備高純氣氣體純化器使用的壽命要求,還需投入大量的工作。
因此,本申請課題擬在前期工作基礎上,針對Au/Fe2O3體系的失活機制,解決制約其在氣體純化器應用的催化劑穩定性問題。通過助劑的選擇和添加對催化劑進行改性是提高催化劑結構穩定性的有效途徑之一。迄今為止,對于Au/Fe2O3催化劑制備工藝的研究多有報道,但針對助劑改善Au/Fe2O3催化劑體系穩定性的研究還不夠系統。
本論文中引入了第五周期過渡金屬、稀土金屬,以考察這些助劑對催化劑性能的影響規律,以期篩選出適宜助劑,使催化劑能保持較高催化活性的同時提高穩定性。并通過對樣品進行X射線粉末衍射(XRD)、比表面積和孔結構測試、程序升溫還原(TPR)表征,以探討助劑對Au/Fe2O3催化劑低溫CO氧化反應性能的調變機理。
1 實驗部分
1.1 Au/Fe2O3-MOx催化劑的制備
將一定濃度的Fe(NO3)3和沉淀劑K2CO3溶液并流加入到攪拌中的底液(pH值為8.0的K2CO3溶液)中,將所得沉淀物經洗滌數次后分散到50 mL去離子水中,并用K2CO3溶液調節pH值為8.0,最后在強烈攪拌下逐滴加入HAuCl4溶液,并用K2CO3溶液維持體系pH值不變,將所得沉淀物洗滌至檢測不到氯離子為止,再經120℃干燥,最后在300℃下焙燒2 h(升溫速率為5℃/min),自然冷卻即制得所需催化劑。
按助劑金屬M與Fe摩爾比為1∶20引入助劑MOx,添加方式為溶液加入法和粉末加入法。其中溶液加入法是將Fe(NO3)3和助劑鹽溶液混合均勻后,采用改性沉積—沉淀法制備樣品。粉末加入法則將一定量的粉末助劑分散到100 mL去離子水中進行超聲波處理10 min后,獲得的乳濁液作為底液,然后將一定濃度的Fe(NO3)3溶液與K2CO3溶液緩慢地并流滴加到底液中,采用改性沉積—沉淀法制備了金含量為0.5%(w/w)(理論添加量)的改性Au/Fe2O3-MOX催化劑。
1.2 催化劑性能的評價
稱取約150 mg、80~100目的催化劑放入反應管中,將反應管裝到管路中,使樣品處于管式爐的恒溫區內進行實驗。原料氣為工業氮,其CO含量為10×10-6;O2含量為100×10-6。
原料氣和反應氣由華愛GC-9560氣相色譜儀的PDHID檢測器測得,最低可檢測到10×10-9,采用兩個氣動六通閥控制Porapak Q和5A分子篩色譜填充柱進樣時間和轉換時間,在60℃下可用于完全分離 H2、O2、N2、CH4、CO 和 CO2,在室溫(25 ℃)連續反應120 h,每隔6 h采集一次數據。
空速計算:
空速=每小時的氣流量(mL·h-1)/[裝填量(g)/密度(g·mL-1)]
接觸時間=裝填量(mg)/每秒氣流量(mL·s-1)
CO的轉化率表示如下:

1.3 催化劑的表征
1.3.1 比表面和孔結構測定
采用NOVA 4200e氣體吸附孔徑測定儀(美國Quantachrome公司)測定催化劑的比表面積和孔結構。樣品在120℃抽真空預處理3~4 h后,以N2為吸附質,在液氮溫度下(77 K)測得吸脫附曲線。用NOVAWin2/2-P Ver.2.1軟件,采用BET法計算比表面積,采用BJH法分析脫附支數據計算孔分布。
1.3.2X射線粉末衍射(XRD)
用瑪瑙研缽將所測樣品研磨成細微顆粒,然后制成平板狀,在荷蘭Panalytic公司X’pert Pro衍射儀上采用 X’Celerator探測器,Co靶 Kα(0.1790 nm)輻射,功率 40 kV×40 mA,掃描步長為0.0333 °,掃描速率為40 s·step-1。
1.3.3 H2-程序升溫還原(H2-TPR)
采用美國Mciromeritics Autochem 2910型自動吸附儀測定催化劑樣品的氧化還原性質。稱取催化劑樣品0.1000 g,置于玻璃反應器內,在120℃下高純氦氣吹掃1 h,降至室溫,繼續用高純氦吹至基線平穩。以10%(V/V)H2/Ar混合氣進行程序升溫還原至700℃,還原氣流30 mL·min-1,升溫速率5℃·min-1。
2 結果與討論
2.1 Au/Fe2O3-MOx催化劑的性能測試
對添加部分第五周期過渡金屬、稀土元素的Au/Fe2O3催化劑進行了常溫(25℃)下120 h的穩定性評價實驗,并與Au/Fe2O3催化劑進行對比,結果如圖1所示。所有的催化劑在常溫下均具有較高的CO氧化反應初始活性(CO轉化率均在100%),但隨著反應時間的延長,Au/Fe2O3催化劑的活性逐漸下降,反應120 h后,CO的轉化率為95.8%,添加La、Mo和Nb對Au/Fe2O3催化劑穩定性影響較小,穩定性變化不明顯,而Ce或Cd的引入則使Au/Fe2O3催化劑穩定性明顯下降,值得注意的是含Zr助劑的樣品穩定性較未改性的Au/Fe2O3催化劑活性明顯改善,經過120 h的反應后,催化劑仍可使CO完全氧化。因此添加ZrO2助劑能有效地提高Au/Fe2O3催化劑的穩定性。

圖1 部分第五周期過渡金屬、稀土元素對Au/Fe2O3催化劑穩定性的影響Fig.1 Effect of promoter from the fifth periodic transition metal or rare earth element on catalytic stability of Au/Fe2O3catalyst
2.2 織構分析

圖2 部分第五周期過渡金屬、稀土元素改性的Au/Fe2O3-MOx催化劑的等溫吸脫附曲線Fig.2 Adsorption-desorption isotherms of Au/Fe2O3 catalysts modified by promoter from the fifth periodic transition metal or rare earth element
圖2和表1分別為對該系列樣品進行N2物理吸附表征獲得的等溫吸脫附曲線及比表面積和孔結構具體的分析數據。該系列助劑的引入對催化劑織構影響相對較小。僅發現助劑Mo和La的引入使樣品比表面積和孔容有所提高,二者的織構性質相近且活性相當;Zr、Nb、Ce的引入依次使催化劑的比表面積略有下降的同時,增大了樣品的孔徑和孔容。同樣地,通過與該系列樣品活性之間關聯,可以確認織構性質并不是決定活性優劣的主要原因。

表1 部分第五周期過渡金屬、稀土元素對Au/Fe2O3-MOx催化劑織構性質的影響Table 1 Effect of promoter from the fifth periodic transition metal or rare earth element on the texture of Au/Fe2O3-MOxcatalysts
2.3 物相組成分析
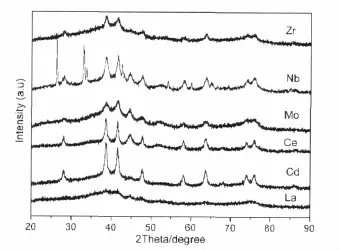
圖3 部分第五周期過渡金屬、稀土元素改性的Au/Fe2O3催化劑的XRD譜圖Fig.3 XRD patters of Au/Fe2O3catalysts modified by promoter from the fifth periodic transition metal or rare earth element
圖3中給出了該系列樣品的XRD譜圖,從中可以明顯看出:(1)引入Zr在一定程度上抑制了載體的晶化,提高了金的分散度,這可能是其改善催化劑性能的主要原因。(2)引入Nb后,載體的晶化程度及金的分散狀況均沒有十分明顯的變化;從譜圖中能清楚地看到峰形較為尖銳的Nb2O5特征衍射峰,這說明通過粉末加入法引入的Nb2O5主要以晶型完好的獨立相存在,其與載體及金的相互作用可以忽略,從而對催化劑的結構和性能影響不大。(3)Mo和La的引入對載體的晶化有明顯的抑制作用,特別是La的引入后載體和金的特征衍射峰均明顯弱化;而Mo的引入對提高金的分散作用不大。(4)Cd的引入對載體晶化影響不大,但明顯提高了金的分散程度;Ce的引入不利于金的分散。通過二者改性后的樣品,活性均急劇下降,我們將在下面進行詳細的探討。
2.4 程序升溫還原(TPR)分析
對該系列樣品進行TPR 表征如圖4所示,發現Ce助劑的引入使得原本處于100℃附近歸屬于表面羥基及無定形氧化鐵還原的峰消失,第二個還原峰明顯增強并向高溫方向移動,由于具有一定活性的載體Fe3O4的形成溫度較高,使得樣品的催化活性有所下降;同時在300℃附近出現了一個可能與表面Ce還原有關的新還原峰;Cd的引入則使第二個還原峰溫向更高溫度移動,相應地該樣品活性大幅度下降;Nb的引入對催化劑還原性能的影響不明顯;而含Zr、Mo和La等助劑的樣品由于載體的晶化程度較低,Au的粒子相對較小,第一個還原峰明顯寬化,第二個還原峰則向低溫方向移動,對應的樣品保持了較高的催化活性。

圖4 第五周期過渡金屬元素改性的Au/Fe2O3催化劑的H2-TPR譜圖Fig.4 H2-TPR profiles of Au/Fe2O3catalysts modified by the fifth periodic transition metal promoter
經助劑改性后,催化劑的性能除了與金和載體的物理化學性質有關外,還與助劑本身的性質及助劑與本體催化劑的相互作用有關。引入Ce和Cd后,使得催化劑的還原能力下降,最終導致催化劑性能下降。因此在所考察的Au/Fe2O3催化體系中Ce、Cd不宜作為改性助劑。La和Mo助劑的引入可較大幅度地提高催化劑的比表面積和孔容,但由于前者改性的催化劑中載體的晶化程度較低,而后者改性的催化劑中Au的粒子尺寸較大,使得二者在低溫反應時,Au與Fe3O4不能較好地協同作用,導致了低溫活性較差。Zr的引入在一定程度上抑制了載體的晶化,并提高了金的分散度,因此具有較高的催化性能。
綜上所述,不同助劑對Au/Fe2O3催化劑的還原性能、金的分散度,載體的結晶度產生影響程度不同,使得改性后的Au/Fe2O3催化劑的CO氧化反應活性明顯不同,還原性能越好、金的分散度越高,以及適宜的載體結晶度有利于催化劑性能的改善。在所考察的助劑中Zr的引入,明顯改善了Au/Fe2O3催化劑的穩定性。
[1]HUSAIN S,KOROS W J.Mixed matrix hollow fiber membranes made with modified HSSZ-1S zeolite in polyetherimide polymer matrix for gas separation [J].Journal of Membrane Science,2007(288):195-207.
[2]MARAND E,PECHAR T W,SANGIL K,et al.Fabrication and characterization of polyimide-zeolite L mixed matrix membranes for gas separations[J].Journal of Membrane Science,2006(277):195-202.
[3]De Vos R M,MAIER W F,VERWEIJ H.Hydrophobic silica membranes for gas separation[J].Journal of Membrane Science,1999(158):277-288.
[4]王書偉,鐘惠安.終端氣體純化技術[J].低溫與特氣,1997,15(1):30-33.
[5]ZALC J M,SOKOLOVSKII V,L?fFLER D G.Are Noble Metal-Based Water-Gas Shift Catalysts Practical for Automotive Fuel Processing[J].Journal of Catalysis,2002(206):169-171.
[6]CAMERONA D,HOLLIDAYB R,THOMPSON D.Gold’s future role in fuel cell systems[J].Journal of Power Sources,2003(118):298-303.
[7]TROVARELLI A,LEITENBURG C,BOARO M,et al.The utilization of ceria in industrials catalysis,Catalysis Today,1999,50(2):353-367.
猜你喜歡
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
中國塑料(2016年12期)2016-06-15 20:30:07
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
中國塑料(2016年5期)2016-04-16 05:25:36
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
