頂空氣相色譜法測定格拉司瓊中的殘留溶劑研究
2013-10-19 02:11:50中國醫科大學附屬第一醫院110001田昕
首都食品與醫藥 2013年10期
中國醫科大學附屬第一醫院(110001)田昕
北京康遠制藥有限公司(100072)劉思飛 趙明 韓程亮Δ
格拉司瓊為選擇性5-HT3受體拮抗劑,主要用于惡性腫瘤化療以及放療過程中的鎮吐及其他相關病癥的治療。格拉司瓊為繼昂丹司瓊上市后的第二個5-HT3受體拮抗劑,與昂丹司瓊相比,格拉司瓊對受體的選擇性更高,止吐作用更強,劑量簡單、作用更為持久。格拉司瓊或其鹽類物質目前的主要給藥途徑為靜脈輸注、口服及外用劑型,現有文獻中未見格拉司瓊殘留溶劑的相關報道。
該產品按照其生產工藝要求,分別使用了甲醇、乙醇、二氯甲烷、異丙醇、乙酸乙酯、四氫呋喃共6種有機溶劑。參照國家食品藥品監督管理局藥品審評中心《化學藥物殘留溶劑研究的技術指導原則》要求,同時參考有關文獻[1][2],并經大量試驗研究,建立了同時測定格拉司瓊中甲醇、乙醇、二氯甲烷等上述6種殘留有機溶劑含量的頂空氣相色譜法。按照ICH及《中國藥典》2010版關于殘留溶劑的有關規定[1][3],制定各溶劑的限度為:本品1g中含甲醇不得超過300μg,乙醇、異丙醇及乙酸乙酯不得超過500μg,二氯甲烷不得超過60μg,四氫呋喃不得超過72μg。根據該原料的溶解特征,筆者采用二甲基亞砜為溶媒,以1,4-二氧六環為內標,采用內標法進行試驗,經方法學考察結果表明各組分的分離度、重現性良好,試驗方法合理,簡單易行,測定結果準確可靠,可作為格拉司瓊中甲醇、乙醇、二氯甲烷、異丙醇、乙酸乙酯、四氫呋喃殘留溶劑的有效測定方法。
1 儀器與試藥
Agilent 6890N氣相色譜儀、Agilent 7689E頂空進樣裝置配有Agilent操作控制及積分軟件;AND HA-202M電子分析天平。
甲醇(美國FISHER公司,批號:076535);乙醇(北京益利精細化學品有限公司,批號:20070728);異丙醇(北京化學試劑公司,批號:070508);二氯甲烷(北京化學試劑公司,批號:40002694);乙酸乙酯(上海試劑一廠,批號:97-03-01);四氫呋喃(北京化學試劑公司,批號:20060628);1,4-二氧六環(北京化工廠,批號:081001);二甲基亞砜(天津市福晨化學試劑廠,批號:20070615、20071105)均為色譜純。
格拉司瓊(批號:080107、080108、080109),由寧波天衡制藥有限公司提供。
內標溶液配制:精密稱取1,4-二氧六環300mg,置于200mL量瓶中,以二甲基亞砜稀釋至刻度,搖勻,作為內標溶液。
對照溶液配制:取色譜純甲醇、乙醇、二氯甲烷、異丙醇、乙酸乙酯、四氫呋喃適量,分別精密稱定,置于同一量瓶中,加內標溶液溶解并稀釋制成每1mL約含甲醇75μg,乙醇、異丙醇、乙酸乙酯125μg,四氫呋喃18μg以及二氯甲烷15μg的均勻溶液作為對照溶液。
2 方法與結果
2.1 色譜條件 采用色譜柱為DB-624交聯毛細管柱(30m×0.53mm.i.d,3μm),以氮氣為載氣,氫火焰離子化檢測器,進樣口溫度為180℃,檢測溫度250℃。柱溫采用程序升溫:40℃保持8min后以10℃/min升至180℃,保持2min;流速為2.5mL/min。頂空進樣(95℃預熱25min),進樣體積為頂空瓶上部氣體1mL,分流比為1∶10。
2.2 分離度試驗 精密量取對照溶液1.0mL于頂空瓶中,按上述方法進行測定,記錄色譜圖;甲醇、乙醇、二氯甲烷、異丙醇、乙酸乙酯、四氫呋喃的分離度均大于1.5,取對照溶液連續進樣6次,所得待測各溶劑峰面積的相對標準偏差均小于5.0%,符合《中國藥典》2010年版二部的相關規定。
2.3 線性范圍及標準曲線 精密稱取甲醇75.0mg、乙醇、異丙醇、乙酸乙酯各125.0mg、二氯甲烷15.0mg及四氫呋喃18.0mg,置于10mL容量瓶中,以內標溶液稀至刻度搖勻,作為標準曲線儲備溶液。精密量取該溶液2.0mL置于10mL容量瓶中加內標溶液稀釋至刻度,搖勻,作為標準曲線溶液。按比例精密稀釋成不同濃度標準溶液6瓶,照上述方法進行測定,以濃度為橫坐標,以各溶劑峰面積與內標峰面積的比值為縱坐標,進行線性回歸,結果見附表1。
2.4 最小檢測限 按照信噪比3∶1,本方法溶液中乙甲醇、乙醇、二氯甲烷、異丙醇、乙酸乙酯、四氫呋喃的最小檢測限分別為0.01μg/mL、0.19μg/mL、0.02μg/mL、0.18μg/m、0.02μg/mL、0.003μg/mL。
2.5 精密度試驗 精密量取對照溶液各1.0mL,置于6個頂空瓶中,照上述方法測定,根據甲醇、乙醇、二氯甲烷、異丙醇、乙酸乙酯、四氫呋喃的峰面積計算RSD%分別為4.25%、4.28%、4.27%、3.33%、3.25%、2.92%,均小于5.0%。

附表1 線性范圍和回歸方程
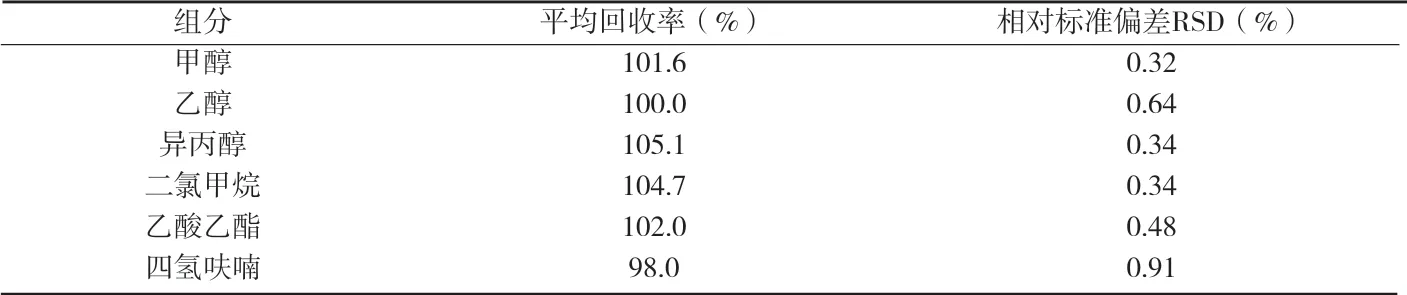
附表2 格拉司瓊的平均回收率
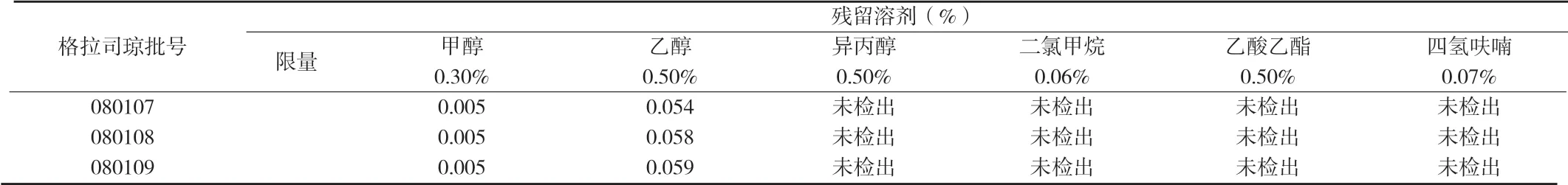
附表3 三批格拉司瓊的殘留溶劑測定結果(%)
2.6 回收率試驗 分別精密稱取格拉司瓊250mg3份于3個頂空瓶中,分別加入對照溶液1mL充分混勻,照上述方法進行測定,通過積分確定其峰面積,按內標法計算試樣中甲醇、乙醇、二氯甲烷、異丙醇、乙酸乙酯、四氫呋喃的平均回收率,結果見附表2。
2.7 樣品測定 精密稱取格拉司瓊3批供試品各約250mg(每批兩份平行),分別于6個頂空瓶中,分別加入內標溶液1.0mL充分混勻,照上述方法進行測定,結果3批供試品中均未檢出異丙醇、二氯甲烷、乙酸乙酯、四氫呋喃等有機溶劑殘留,甲醇和乙醇殘留量均符合規定,結果見附表3。
3 討論
3.1 預熱溫度及時間的選擇 本試驗根據樣品的熱穩定性以及溶解度分別選擇于75、85、95及105℃條件下預熱,經過比較在溫度相對較低的條件下,樣品的回收率較低,而過高的溫度對于樣品的穩定性以及低沸點的溶劑也會造成一定影響;而后,筆者對預熱時間進行了選擇,分別預熱15、20、25、30min后進行測定,最終選擇了95℃、預熱25min作為預熱條件。
3.2 內標物質的選擇 氣相色譜的測定過程中,使用外標法定量容易造成多針進樣、重現性不好、測定結果不準確等問題,故采用內標法對其進行定量。經過試驗,分別考察了乙醇、丙酮以及1,4-二氧六環等多種內標物質,結果發現在該條件下1,4-二氧六環的出峰時間與其他溶劑出峰無干擾,最終確定1,4-二氧六環為本試驗中的內標物質。
3.3 關于格拉司瓊殘留溶劑的測定 有文獻報道[4],鹽酸格拉司瓊中的殘留溶劑測定方法,其采用外標法測定鹽酸格拉司瓊中的甲醇、異丙醇、乙醚、甲苯等8種有機溶劑。鹽酸格拉司瓊為格拉司瓊鹽酸鹽形式,溶解特征與格拉司瓊有較大差異,主要用于靜脈給藥的注射液中;在口服以及外用等給藥途徑中,則主要以格拉司瓊原型作為原料制成相應制劑。本文建立的格拉司瓊中殘留溶劑的測定方法能夠同時測定生產工藝中涉及的6種有機溶劑的殘留,所建立的方法準確、可靠,可用于格拉司瓊中殘留溶劑的測定,保障藥品的安全、有效。
