超高效液相色譜串聯(lián)質(zhì)譜法測定水體與底泥中孔雀石綠及隱色孔雀石綠殘留
2013-11-28 01:00:26孫言春李池陶杜寧寧曹頂臣牟振波王海濤陳中祥
分析測試學(xué)報(bào) 2013年2期
關(guān)鍵詞:方法
孫言春,李池陶,杜寧寧,曹頂臣,牟振波*,吳 松,王海濤,陳中祥
(1.中國水產(chǎn)科學(xué)研究院 黑龍江水產(chǎn)研究所/農(nóng)業(yè)部水產(chǎn)品質(zhì)量安全風(fēng)險(xiǎn)評估實(shí)驗(yàn)室(哈爾濱),黑龍江 哈爾濱 150070;2.哈爾濱工業(yè)大學(xué) 理學(xué)院,黑龍江 哈爾濱 150001)
孔雀石綠(Malachite green,MG)又名堿性綠、苯胺綠,曾作為一種工業(yè)染料廣泛用作羊毛、絲綢、皮革等的染色劑[1]。由于MG還具有殺滅真菌、寄生蟲等病原微生物的作用,在水產(chǎn)養(yǎng)殖中常用來預(yù)防和治療受精卵和成魚的水霉病、腮霉病和小瓜蟲病等,以及用于活魚運(yùn)輸、池塘?xí)吼B(yǎng)過程和環(huán)境的消毒等[2-5]。然而,MG在生物體內(nèi)有明顯蓄積現(xiàn)象且具有高毒、高殘留、致癌、致畸、致突變等毒副作用,對生物體的組織、生殖、免疫系統(tǒng)均有影響,尤其是其代謝產(chǎn)物隱色孔雀石綠(Leucomalachite green,LMG)毒性更大[6-12]。因此,包括歐美、中國在內(nèi)的國家都明令禁止其在水產(chǎn)養(yǎng)殖中的應(yīng)用[13],以確保消費(fèi)者健康和水產(chǎn)貿(mào)易的對外地位。
雖然MG在水產(chǎn)養(yǎng)殖中的違禁使用情況越來越少,但由于其曾經(jīng)被廣泛應(yīng)用,已對環(huán)境造成了嚴(yán)重污染,尤其是表層沉積物作為MG在環(huán)境中的最終歸宿,且不斷向水體進(jìn)行緩慢釋放,成為其二次污染的主要來源[5,14-15]。因此,評估表層沉積物和水體中的MG及其代謝物殘留狀況,建立相應(yīng)的分析方法,對于水產(chǎn)品中MG污染來源的排查具有積極的意義。
目前,針對水體、底泥等環(huán)境中的痕量MG及其代謝物的檢測方法相對較少,且主要采用靈敏度較低的液相色譜法[16-19]。由于底泥等沉積物的成份復(fù)雜,各種雜質(zhì)多,對分析干擾大,高效液相色譜對樣品的凈化要求較高,整個(gè)操作步驟繁瑣、耗時(shí)長,不利于準(zhǔn)確定性和定量分析。而高效液相色譜串聯(lián)質(zhì)譜法由于靈敏度高、專屬性強(qiáng),因此在MG藥物殘留檢測上獲得了廣泛的應(yīng)用[20-23]。已有研究采用高效液相色譜串聯(lián)質(zhì)譜法測定了水或底泥中MG和隱色LMG殘留[24-26],但其前處理方法較繁瑣,不適合大批量樣品的檢測需要。
本研究充分利用高效液相色譜串聯(lián)質(zhì)譜靈敏度高、專屬性強(qiáng)的優(yōu)點(diǎn),對水體及表層沉積物中的MG及其代謝物進(jìn)行提取、濃縮,優(yōu)化了前處理過程,能夠快速準(zhǔn)確檢測出養(yǎng)殖水體及沉積物中殘存的痕量MG及其代謝物,可為污染來源排查及評價(jià)生態(tài)環(huán)境對健康養(yǎng)殖的影響提供可靠的檢測手段。
1 實(shí)驗(yàn)部分
1.1 儀器設(shè)備
AcquityTM超高效液相色譜儀,Micromass Quattro micro API三重四極桿質(zhì)譜(配電噴霧離子源),MassLynxTMV 4.1操作軟件(Waters公司);XS205電子天平(Mettler Toledo公司);GX271自動固相萃取儀(Gilson公司);Biofuge Stratos高速冷凍離心機(jī)(Thermo Scientific公司);T25 digital Ultra-Turrax均質(zhì)機(jī),MS1 Mini-shaker渦輪振蕩器(IKA公司);Milli-Q A10純水器(Millipore公司);移液器(200、1 000、5 000 μL,Eppendorf公司);PHS-3C pH計(jì)(上海精密科學(xué)儀器有限公司)。
1.2 試劑與材料
MG、LMG、MG-D5、LMG-D6(純度≥99.5%,德國Dr.Ehrenstorfer公司);甲醇、乙腈(色譜純,德國Merck公司);二氯甲烷(色譜純,美國Fluka公司);實(shí)驗(yàn)用水由Millipore A10純水系統(tǒng)(美國Millipore公司)制備;乙酸銨(LC/MS級)、乙酸(色譜純)、甲酸(純度>98%)均購自美國Fluka公司;N,N,N',N'-四甲基對苯二胺(TMPD,生物試劑),濃鹽酸(分析純);固相提取小柱Agilent Accu-BOND SCX(美國Agilent公司,200 mg/3 mL),Oasis MCX(美國Waters公司,150 mg/6 mL),Oasis WCX(美國Waters公司,150 mg/6 mL)。
TMPD溶液(1.0 g/L):稱取50 mg TMPD溶于50 mL甲醇中;檸檬酸緩沖溶液(pH 2.6):10.5 g檸檬酸鹽溶入500 mL水中,配成0.1 mol/L檸檬酸鹽溶液;將14.2 g磷酸二氫鈉(NaH2PO4·2H2O)溶于500 mL水中,配成0.2 mol/L磷酸二氫鈉溶液,將445.5 mL 0.1 mol/L檸檬酸鹽溶液和54.5 mL 0.2 mol/L磷酸二氫鈉溶液混合即可;標(biāo)準(zhǔn)溶液:準(zhǔn)確稱取MG和LMG標(biāo)準(zhǔn)品及對應(yīng)內(nèi)標(biāo),用甲醇溶解并稀釋成100 mg/L的儲備液,室溫下可穩(wěn)定3個(gè)月;儲備液用80%乙腈溶液(乙腈∶5 mmol/L的乙酸銨溶液=8∶2,體積比)作稀釋液配成1.0 mg/L的中間工作溶液,然后用該稀釋液將中間液配制成相應(yīng)濃度的標(biāo)準(zhǔn)工作溶液;內(nèi)標(biāo)溶液使用前用稀釋液配成兩種物質(zhì)濃度均為0.1 mg/L的混合溶液。
1.3 采樣方法
水樣采集:用廣口瓶取500 mL水樣,加入40 mL TMPD溶液,4 000 r/min離心5 min,超聲3 min,過0.45 μm尼龍濾膜。加入100 μg/L MG及LMG內(nèi)標(biāo)混合液50 μL,放置10 min,然后加入檸檬酸緩沖溶液(pH 2.6)和甲醇各30 mL。超聲10 min后,進(jìn)行固相萃取。
底泥樣品采集:取表層沉積物樣品,用均質(zhì)機(jī)充分均質(zhì)后,稱取5.0 g,加入200 μL TMPD溶液和50 μL 100 μg/L的MG及LMG內(nèi)標(biāo)混合液,放置10 min,備用。
1.4 色譜條件
色譜柱:Waters Acquity UPLC BEH C18(1.7 μm,50 mm×2.1 mm);柱溫:35℃;樣品室溫度:10℃;流動相A:含0.1%甲酸的5 mmol/L的乙酸銨水溶液;流動相B:0.1%甲酸乙腈溶液;進(jìn)樣體積:10 μL;流速:0.30 mL/min。梯度洗脫程序:0~1.0 min,40%B;1.0~3.0 min,40%~95%B;3.0~5.0 min,95%~40%B。
1.5 質(zhì)譜條件
電離方式:ESI(+);電離電壓:2.5 kV;錐孔電壓:45 V;離子源溫度:110℃;錐孔反吹氣流量:50 L/h;脫溶劑氣溫度:380℃;脫溶劑氣流量:550 L/h;采集方式:多反應(yīng)監(jiān)測(MRM);定性、定量離子對見表1。
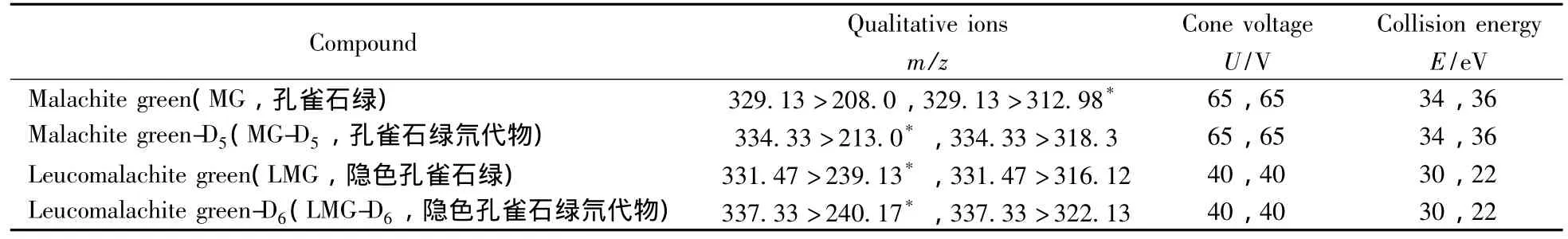
表1 目標(biāo)化合物的MRM監(jiān)測離子對及其質(zhì)譜參數(shù)Table 1 Mass spectrometric conditions for monitoring of target compounds
1.6 前處理方法
水樣:預(yù)先用5 mL甲醇、5 mL水、5 mL檸檬酸緩沖溶液(pH 2.6)活化Waters Oasis MCX(150 mg,6 mL)SPE柱,然后將“1.3”制備的水樣過柱。依次用3 mL 0.1 mol/L的鹽酸溶液、5 mL水、5 mL 50%甲醇水溶液、3 mL正己烷淋洗,棄去淋洗液。真空抽干后,用5 mL乙酸乙酯-甲醇-氨水(50∶45∶5)混合溶液進(jìn)行洗脫,收集于10 mL離心管中,于45℃下氮?dú)獯蹈伞埩粑锛尤?.0 mL的乙腈-5 mmol/L乙酸銨溶液(80∶20),渦旋振蕩1 min,過0.22 μm有機(jī)相濾膜后,上機(jī)檢測。
底泥樣品:準(zhǔn)確稱取(5.0±0.05)g底泥樣品于50 mL聚乙烯離心管中,加入15 mL乙腈,旋渦2 min,超聲萃取20 min后靜置30 min;再向離心管中加入10 mL二氯甲烷,旋渦2 min后,4 000 r/min離心5 min,取下層清液于旋轉(zhuǎn)蒸發(fā)瓶;再次向離心管中加入10 mL乙腈-二氯甲烷混合液(3∶2),重復(fù)上述操作,合并萃取液,于45℃旋轉(zhuǎn)蒸干;殘余物加入1.0 mL的乙腈-5 mmol/L乙酸銨溶液(80∶20),渦旋振蕩1 min,過0.22 μm有機(jī)相濾膜后,上機(jī)檢測。
2 結(jié)果與討論
2.1 液相色譜及質(zhì)譜條件的優(yōu)化
MG及其代謝物L(fēng)MG屬于三苯基甲烷類化合物,具有弱堿性,因此可以通過調(diào)節(jié)流動相的pH值來抑制弱堿的解離,同時(shí)加入甲酸和乙酸銨有利于目標(biāo)分析物的分子離子化,并改善峰形。采用梯度洗脫有利于將目標(biāo)分析物和基質(zhì)分開,并有利于提高離子化效果。在已有文獻(xiàn)的研究基礎(chǔ)上[17-18,21],通過優(yōu)化得到的液相洗脫條件如“1.4”所示。根據(jù)MG和LMG的化學(xué)性質(zhì),選擇ESI正離子模式。用流動注射進(jìn)樣方式對MG、LMG及其對應(yīng)內(nèi)標(biāo)物質(zhì)的母離子進(jìn)行掃描,得到其準(zhǔn)分子離子峰,再以適當(dāng)?shù)呐鲎材芰繉?zhǔn)分子離子進(jìn)行子離子掃描,響應(yīng)值最強(qiáng)的子離子作為定量離子,響應(yīng)值相對較強(qiáng)的離子作為定性離子。以MRM掃描模式對母離子與子離子進(jìn)行碎裂電壓及碰撞能量的優(yōu)化,優(yōu)化后的參數(shù)見表1。采用優(yōu)化后的色譜條件和質(zhì)譜條件,MG和LMG及其對應(yīng)內(nèi)標(biāo)的標(biāo)準(zhǔn)溶液質(zhì)量色譜圖如圖1所示。
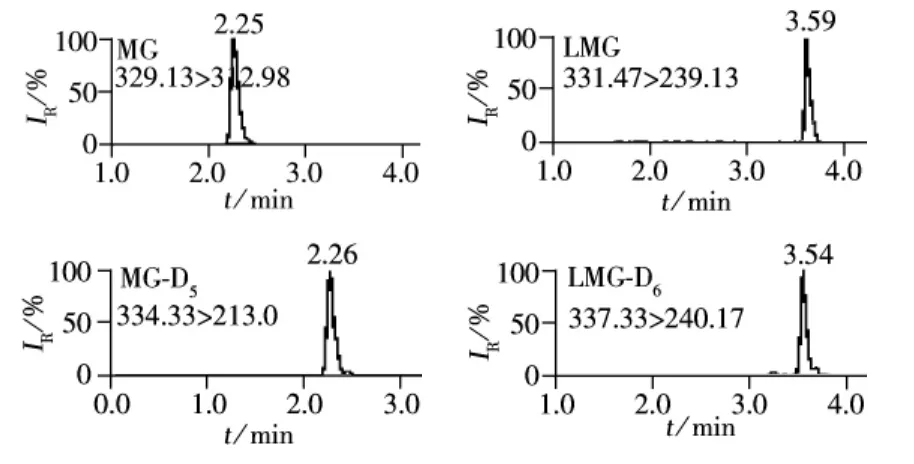
圖1 MG、LMG及其對應(yīng)內(nèi)標(biāo)標(biāo)準(zhǔn)品溶液(2.0 μg/L)的 MRM 色譜圖Fig.1 MRM chromatograms of MG,LMG ,MG-D5,LMG-D6at 2.0 μg/L standard solution
2.2 水溶液萃取固相萃取柱的選擇
水體中MG和LMG含量一般較低,因此需對樣品進(jìn)行提取、富集和凈化。現(xiàn)有方法中,生物基質(zhì)多采用氧化鋁柱凈化,但水體極性較大,氧化鋁對目標(biāo)化合物有一定的吸附造成目標(biāo)化合物回收率偏低。因此,本文比較了Oasis MCX、Oasis WCX和AccuBOND SCX 3種陽離子交換固相萃取柱對水體中MG和LMG的凈化效果,發(fā)現(xiàn)MCX柱對這2種化合物的保留效果較好(見表2)。WCX柱對 MG保留較差,回收率低,對LMG保留較好;而SCX柱的效果則相反。這可能是因?yàn)镺asis MCX柱具有反相與陽離子交換雙重機(jī)理,其選擇性更強(qiáng),效果更好,因此本研究選用Oasis MCX柱進(jìn)行樣品凈化處理。考慮到水環(huán)境中MG及其代謝物的濃度一般較小,按照最大吸附量是填料的二十分之一來計(jì)算,選擇150 mg/6 mL規(guī)格的固相萃取柱已滿足分析方法的要求。

表2 WCX、MCX和SCX固相萃取柱對回收率的影響Table 2 Influence of WCX,MCX and SCX SPE on recoveries of MG and LMG
2.3 固相萃取條件的優(yōu)化
陽離子交換柱適合堿性化合物的富集和凈化,因此一般采用酸性溶液活化,堿性溶液洗脫。本實(shí)驗(yàn)在甲醇和水活化的基礎(chǔ)上,選用pH 2.6的檸檬酸緩沖溶液繼續(xù)活化,可保持填料對陽離子和非極性分子的吸附活性,同時(shí)使目標(biāo)分析物保持正離子態(tài)而被吸附保留在柱上,從而提高填料對目標(biāo)分析物的吸附活性。由于需要堿性有機(jī)溶液才能使填料失活解吸,因此可以選擇用酸性或中性溶液淋洗萃取柱,洗去極性大和極性較小的共存有機(jī)干擾物質(zhì),最后用堿性溶液洗脫。本實(shí)驗(yàn)按照極性逐漸減小的順序,依次用0.1 mol/L的鹽酸溶液、水、50%甲醇水溶液、3 mL正己烷淋洗,以去除吸附在填料上的大部分基質(zhì)干擾。同時(shí)考察了甲醇-氨水(95∶5)、乙腈-氨水(95∶5)、乙酸乙酯-甲醇-氨水(50∶45∶5)3種洗脫液的洗脫效果。結(jié)果表明,在同等體積下,乙酸乙酯-甲醇-氨水(50∶45∶5)的洗脫效果最好,回收率最高,因此選擇其為洗脫溶液。將500 mL 5.0 μg/L的水溶液按照“1.6”方法過柱后,持續(xù)加入12 mL洗脫液,分別用過濾管收集流出液,每2 mL收集1管。45℃下氮?dú)獯蹈珊螅瑲堄辔锛尤?.0 mL乙腈-5 mmol/L的乙酸銨溶液(80∶20),渦旋振蕩1 min,過0.22 μm有機(jī)相濾膜后,上機(jī)檢測。發(fā)現(xiàn)8 mL以后的洗脫液中,均檢測不到目標(biāo)分析物,說明8 mL洗脫液可將目標(biāo)物完全洗脫。為保證洗脫完全,在實(shí)際應(yīng)用時(shí)采用10 mL洗脫液進(jìn)行洗脫。濃縮富集前加入MG-D5和LMG-D6內(nèi)標(biāo),結(jié)果顯示方法的回收率、穩(wěn)定性和重現(xiàn)性均較好。
2.4 底泥樣品中濃縮富集條件的優(yōu)化
底泥樣品均質(zhì)稱量后,需加入一定量的TMPD溶液作為抗氧化劑來提高樣品中MG和LMG的回收率。對固體樣品中MG和LMG的提取,常用的提取溶劑包括乙腈、甲醇、乙酸銨緩沖液和二氯甲烷等[18-23]。常用的方法是先加入乙腈,使底泥中的目標(biāo)分析物溶解到乙腈溶液中,然后用二氯甲烷將乙腈中的MG和LMG反萃取到二氯甲烷溶液中,最后將二氯甲烷提取液進(jìn)一步濃縮和凈化。本實(shí)驗(yàn)考察了乙腈和二氯甲烷的體積比分別為2∶3、1∶1和3∶2時(shí)的提取效果,回收率結(jié)果如表3所示。結(jié)果表明,乙腈與二氯甲烷采用3∶2比例時(shí),回收率最高,穩(wěn)定性較好,因此選擇乙腈和二氯甲烷體積比為3∶2的混合液為提取液。以有機(jī)試劑對固體粉碎樣品進(jìn)行提取時(shí),一般采用有機(jī)試劑的體積為固體樣品質(zhì)量的5倍左右,以使提取液和樣品表面充分接觸而提高提取效果,因此選擇加入25 mL提取液,即先加入15 mL乙腈,渦旋和超聲提取后,再加入10 mL二氯甲烷渦旋和離心。再次向離心管中加入10 mL乙腈-二氯甲烷混合液提取液,重復(fù)提取一次,合并萃取液,于45℃旋轉(zhuǎn)蒸干,可使回收率達(dá)到80%以上。
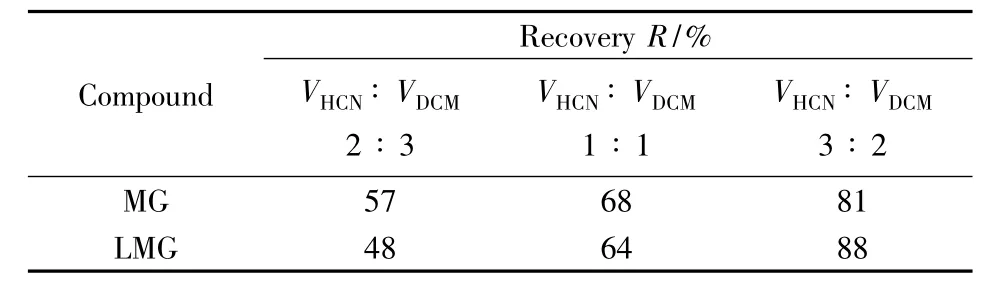
表3 不同乙腈和二氯甲烷體積比對回收率的影響Table 3 Influence of different volume ratio of HCN to DCM on recoveries of MG and LMG
2.5 方法的標(biāo)準(zhǔn)曲線與靈敏度
在優(yōu)化條件下,分別采用內(nèi)標(biāo)法和外標(biāo)法考察了MG和LMG的線性范圍(見表4),結(jié)果顯示兩者質(zhì)量濃度均在0.2~100 μg/L范圍內(nèi)與峰面積呈良好的線性關(guān)系,r2大于0.995。但采用外標(biāo)法對實(shí)際樣品進(jìn)行定量時(shí),MG和LMG在環(huán)境水和底泥樣品中的回收率分別在45%和60%左右,這可能是基質(zhì)效應(yīng)造成的結(jié)果。而采用內(nèi)標(biāo)法定量時(shí),回收率分別在80%和90%左右,大大減少了基質(zhì)效應(yīng)帶來的影響,因此本方法采用內(nèi)標(biāo)法定量。
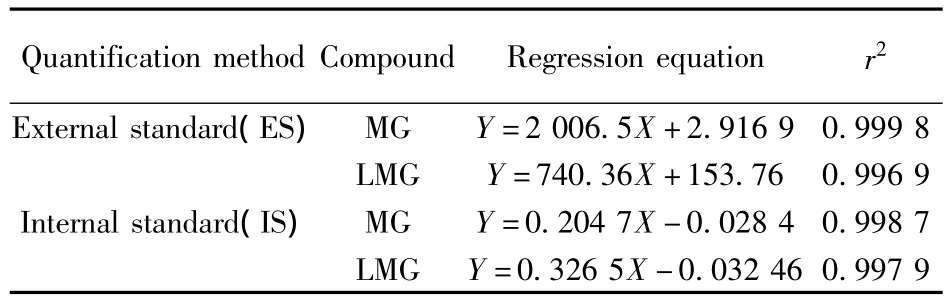
表4 MG和LMG的回歸方程及相關(guān)系數(shù)(r2)Table 4 Regression equation and correlation coefficients(r2)of MG and LMG
采用向陰性樣品中添加標(biāo)準(zhǔn)物質(zhì)的方法,進(jìn)一步考察方法的靈敏度。在方法規(guī)定的取樣體積和定容體積下,按3倍信噪比計(jì)算得該方法在環(huán)境水樣和底泥樣品中的檢出限(LOD)分別為0.2 ng/L和0.02 μg/kg,以信噪比S/N>10得其定量下限(LOQ)分別為0.4 ng/L和0.04 μg/kg。
2.6 方法的回收率與精密度
采用空白加標(biāo)的方式,在環(huán)境水樣中分別添加1.0、10.0、100 ng/L 3個(gè)濃度梯度,底泥中MG和LMG分別添加0.20、2.00、20.0 μg/kg,按照本方法處理后測定,回收率和相對標(biāo)準(zhǔn)偏差見表5。
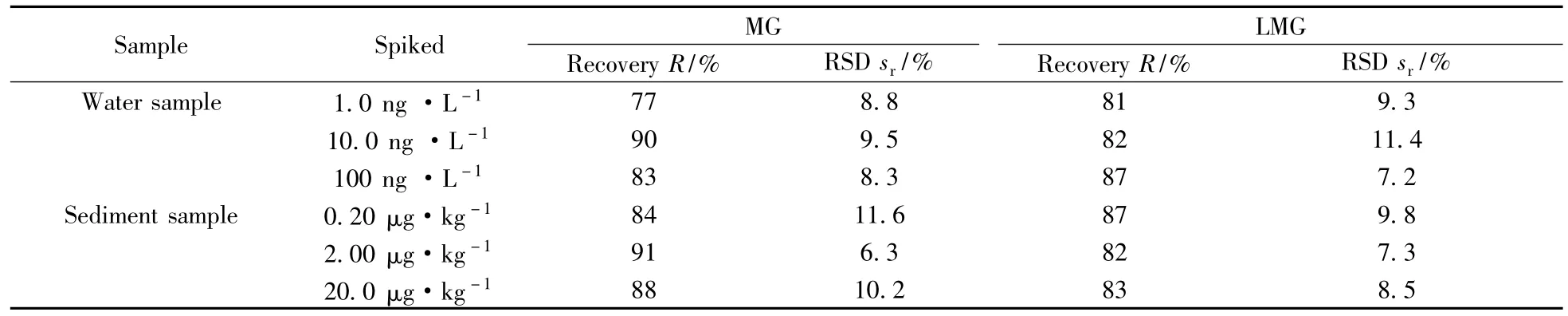
表5 養(yǎng)殖環(huán)境水和底泥中MG和LMG的加標(biāo)回收率及相對標(biāo)準(zhǔn)偏差Table 5 Spiked recoveries and RSDs of MG and LMG in negative water and sediment in aquatic environment
由表5可見,在養(yǎng)殖環(huán)境水體中,MG和LMG的平均回收率為77%~90%,RSD為7.2%~11.4%;底泥中MG和LMG的平均回收率為82%~91%,RSD為6.3%~11.6%。因此,MG和LMG在水產(chǎn)養(yǎng)殖環(huán)境水和底泥中的加標(biāo)回收率均能達(dá)到檢測要求,說明本方法具有較高的準(zhǔn)確性。同時(shí),RSD均低于15%,表明結(jié)果在置信區(qū)間內(nèi),本方法具有較高的可靠性。
2.7 實(shí)際樣品的測定
采用該方法測定了東北地區(qū)11家水產(chǎn)養(yǎng)殖場的養(yǎng)殖池塘水體和底泥中MG和LMG的殘留情況,在3家檢出的養(yǎng)殖場中,其水體中MG的濃度為2.7~11.6 ng/L,LMG的濃度為6.5~21.6 ng/L;底泥中MG的濃度為0.8~18.7 μg/kg,LMG的濃度為2.8~26.4 μg/kg。經(jīng)進(jìn)一步調(diào)查,發(fā)現(xiàn)該3家養(yǎng)殖場在幾年前確曾采用過MG來防治和治療水霉病,因此,該方法可用于水產(chǎn)養(yǎng)殖環(huán)境水體和底泥中MG和LMG殘留量的測定。
3 結(jié)論
本文通過優(yōu)化固相萃取條件和液液萃取條件,建立了水產(chǎn)養(yǎng)殖環(huán)境水體和底泥中MG和LMG殘留的高靈敏分析和確證方法。結(jié)果表明,本方法在0.2~100 μg/L范圍內(nèi)呈現(xiàn)良好的線性關(guān)系,水體和底泥中的檢出限分別為0.2 ng/L和0.02 μg/kg;相對標(biāo)準(zhǔn)偏差(RSD)均小于12%。該方法步驟簡單、回收率高、精密度良好。可作為水體和底泥中MG及其代謝物L(fēng)MG殘留的檢測方法,并可為MG及LMG在水產(chǎn)品中的富集規(guī)律及毒理評價(jià)提供靈敏、準(zhǔn)確的分析手段,為水產(chǎn)品中MG及LMG溯源和衛(wèi)生監(jiān)督提供檢測技術(shù)支持。
[1]Culp S J,Beland F A.Int.J.Toxicol.,1996,15(3):219 -238.
[2]Li C T,Xu W,Jia Z Y,Shi L Y.Chin.J.Fish.(李池陶,徐偉,賈智英,石連玉.水產(chǎn)學(xué)雜志),2011,24(4):37-40.
[3]Eli A,Briyai O F,Abowei J F N.Asian J.Med.Sci.,2011,3(5):198-205.
[4]Ni D S.Research of Prevent and Control Saprolegniasis in Fishes.Beijing:Agriculture Press(倪達(dá)書.魚類水霉病防治研究.北京:農(nóng)業(yè)出版社),1982:32.
[5]Alderman D J.J.Fish.Dis.,1985,8(3):289 -298.
[6]Shivaji S,Ranjana S,Roy D.Aquat.Toxicol.,2004,66(3):319 -329.
[7]Hou Y Q,Zhu W J.Reproduction and Contraception(侯瑜瓊,朱偉杰.生殖與避孕),2006,26(12):707-711.
[8]Zhou Y H,Wu Y K,Li Q X,Zhang J X,Shen J Z,Hu Z M.J.Toxicol.(周艷紅,吳永魁,李乾學(xué),張錦霞,沈建忠,胡仲明.毒理學(xué)雜志),2006,20(3):163-165.
[9]Wen C,Wu H J,Wang Y F.Environ.Sci.Technol.(文琛,鄔紅娟,王宇飛.環(huán)境科學(xué)與技術(shù)),2007,30(6):8-9.
[10]Li N.Foreign Med.Sci.:Sect.Hyg.(李寧.國外醫(yī)學(xué) 衛(wèi)生學(xué)分冊),2005,32(5):262-264.
[11]Sandra J C,Lonnie R B,Donna F K,Daniel R D,Louis T M,F(xiàn)rederick A B.Chem.Biol.Interact.,1999,122(1):153-170.
[12]Annalaura S,Carlo N,Isabella D A,Alessandra G A,Monica C,Claudia R,F(xiàn)ranco Z,Mauro D.Toxicol.Vitro.,2005,19(7):853-858.
[13]Sudova E,Machova J,Svobodova Z,Vesely T.Veter.Med.,2007,52(12):527 -539.
[14]Kumar K V,Sivanesan S,Ramamurthi V.Process Biochem.,2005,40(8):2865-2872.
[15]Gupta V K,Mittal A,Krishnan L,Gajbe V.Sep.Purif.Technol.,2004,40(1):87 -96.
[16]Safarik I,Safariková M.Water Res.,2002,36(1):196 -200.
[17]Zhang T Q,Yang H S,Lin H,Wang J,Zhou G,Li Q.Journal of Fisheries of China(張彤晴,楊洪生,林海,王靜,周剛,李強(qiáng).水產(chǎn)學(xué)報(bào)),2007,31(5):699-703.
[18]Yu P J.Chem.Anal.Meter.(余培健.化學(xué)計(jì)量分析),2007,16(2):27-29.
[19]Gao L,Zhang D,Cao J,Guo D,Yang R Z,Chen Y.J.Instrum.Anal.(高玲,張丹,曹軍,郭棟,楊瑞章,陳勇.分析測試學(xué)報(bào)),2012,31(3):337-342.
[20]Kirsi H,Erja L,Kimmo P.J.Chromatogr.B,2007,845(1):74-79.
[21]Zhu K Z,Wang P,Lin Y F,Xiao S J,Mei S R.Chin.J.Chromatogr.(朱寬正,王鵬,林雁飛,蕭松建,梅素容.色譜),2007,25(1):66-69.
[22]María J M B,Sonia H,Ana U,Ana A,María D H,Olga S,Marcus R,Amadeo R F A.Anal.Chim.Acta,2010,665(1):54-74.
[23]Luis V,Cecilia D,Antonio L Z,Pablo R.J.Chromatogr.A,2005,1067(1/2):101-105.
[24]Xu Y H,Gu L,Jiang J S,Ding L.Chem.Anal.Meter.(徐彥輝,顧亮,蔣俊樹,丁磊.化學(xué)計(jì)量分析),2011,20(2):49-51.
[25]Liu G H,Liu H H.J.Hyg.Res.(劉桂華,劉紅河.衛(wèi)生研究),2007,36(6):731-733.
[26]Na G S,Li H X,Zhou C G,Sun Q,Yao Z W.Chin.J.Anal.Lab.(那光水,李紅霞,周傳光,孫茜,姚子偉.分析試驗(yàn)室),2008,27(12):63-65.
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(bào)(2021年2期)2021-05-25 02:07:46
中學(xué)生數(shù)理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(bào)(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
