石墨烯-氧化鎢復合薄膜的制備及界面電子傳輸特性
2014-02-18 12:06:54李文章楊亞輝陳啟元
物理化學學報 2014年10期
李文章 劉 洋 李 潔 楊亞輝 陳啟元
(1湖南農業大學資源環境學院,長沙410128;2中南大學化學化工學院,長沙410083)
1 引言
太陽能作為可再生能源,利用其制氫將有望解決目前較為普遍的能源匱乏及環境污染問題,因而受到了人們的廣泛關注.在眾多的半導體光催化制氫材料中,氧化鎢因性能穩定、成本低廉等特點成為目前光催化領域的熱點研究材料,但其光生電子-空穴易復合限制了器件的光電性能.為了提高氧化鎢的光電轉化性能,國內外的研究人員進行了許多的探索,目前常用的方法有半導體復合、1-3貴金屬沉積、4-6表面敏化、7離子摻雜8,9等.
近年來的研究發現,還原氧化石墨烯(RGO)作為電子傳遞介質,可提高半導體材料中光生電子的遷移速率,降低光生電子-空穴的復合幾率,從而提高半導體材料的光電轉化效率.與碳納米管(CNTs)和富勒烯(C60)相比,石墨烯具有更大的比表面積(2630 m2?g-1)、較高的化學穩定性以及更為優異的電子傳導性能.10,11同時,利用石墨烯規整的二維平面結構作為載體,將石墨烯與半導體催化劑復合,不僅可以提高光生電荷的遷移速率,還可以提高催化劑的分散程度以及復合材料的光催化活性.12-14Guo等15以磷鎢酸和氧化石墨烯為原料,超聲混合后,經熱分解處理制得WO3/石墨烯復合物,與純的WO3體系相比,光解水性能提升了1倍.Yu等16以Na2WO4?2H2O為原料,180°C水熱制備棒狀WO3-石墨烯復合物,光降解和氣敏性能分別提升了1.2和1.5倍.Yang等17以磷鎢酸為鎢源,三嵌段共聚物P123為模板,合成了具有規整結構的介孔WO3-石墨烯復合物,光解水析氧性能提升了4.1倍.Zheng等18則采用水熱法在導電玻璃(FTO)上制備了片狀形貌的氧化鎢-石墨烯復合物,光電性能具有顯著提升,光電流達到2.0 mA?cm-2(1.23 Vvs.RHE).
目前,對負載在導電基底上的WO3-石墨烯復合薄膜的研究還比較少,對其性能提升的原因也都歸結于光生電荷傳輸性能的提升和界面電阻的下降,而關于石墨烯改性后材料的界面電子轉移行為還不十分清楚.基于此,本文采用液相法制備了石墨烯-WO3復合結構薄膜,利用交流阻抗、瞬態光電流譜和強度調制光電流譜等方法,研究復合薄膜電極在光電作用下界面上的載流子轉移過程和電荷傳輸行為,以期能進一步闡明石墨烯對復合結構薄膜的光電性能影響機制.
2 實驗部分
2.1 試劑與儀器
偏鎢酸銨(AMT,(NH4)6W7O24?6H2O,AR,國藥集團化學試劑有限公司),石墨粉(CP,325目,天津市富宇精細化工公司),P2O5(AR,上海凌峰化學試劑有限公司),K2S2O8(AR,天津市光復科技發展有限公司),NaNO3(AR,國藥集團化學試劑有限公司),KMnO4(AR,臺山市化工廠有限公司),聚乙烯吡咯烷酮(PVP,AR,國藥集團化學試劑有限公司),聚乙二醇1000(PEG1000,AR,天津市光復精細化工研究所).
場發射掃描電子顯微鏡(美國FEI公司,Nova Nano SEM 230);透射電子顯微鏡(美國FEI公司,TECNAI G2 F20);X-射線衍射儀(日本Rigaku公司,D/max2250);紫外-可見共焦顯微拉曼光譜儀(英國Renishow公司,UV-1000);電化學工作站(德國Zahner公司,Zennium);CIMPS光電化學譜儀(德國Zahner公司).
2.2 石墨烯-WO3復合薄膜的制備
以改進Hummer法19制備石墨烯的前驅體——氧化石墨烯(GO),將GO超聲分散于去離子水中.將2.0 g PVP溶解于15.0 mL去離子水中,攪拌的同時加入超聲分散后的GO分散液.向體系中加入7.5 g AMT,攪拌4 h.再向溶液中加入1.0 g PEG1000,室溫下攪拌2 h.采用浸置提拉法制備薄膜,以44 mm?min-1的速度將預處理過的摻雜氟的SnO2(FTO)透明導電玻璃從溶液中提拉出來,室溫下干燥.以2°C?min-1速率升溫,450°C熱處理2 h,自然冷卻后得到石墨烯-WO3復合物(G-WNC);以同樣的方法,不加入GO前驅體制備WO3納米晶(WNC)薄膜.
2.3 石墨烯-WO3復合薄膜的光電性能檢測
采用三電極體系對樣品進行光電化學性能測試.以所制備的薄膜器件為工作電極,Pt電極為對電極,Ag/AgCl(飽和KCl)電極為參比電極,0.5 mol?L-1的H2SO4溶液為測試電解質.采用500 W氙燈平行光光源(400 nm紫外濾光片濾掉紫外光),激發光透過石英玻璃從背面照射到薄膜電極表面,光源照射到薄膜表面的光照強度約為100 mW?cm-2.
光電流密度-電位(I-V)測試條件為:電位范圍為0-1.6 V(vsAg/AgCl),掃描速率為20 mV?s-1;交流阻抗譜測試條件為:在0.8 V(vsAg/AgCl)電位下,采用幅值為10 mV的擾動電位,掃描頻率范圍為0.1-10 kHz;瞬態光電流譜測試:電位范圍為0.5-1.2 V(vsAg/AgCl),取樣點為0.5 s-1,分別測試電流與時間的關系.強度調制光電流譜(IMPS)測試:采用PP210驅動的藍色發光二極管提供光源,正弦擾動光強為直流光強的10%,頻率范圍為3 kHz-0.1 Hz.
3 結果與討論
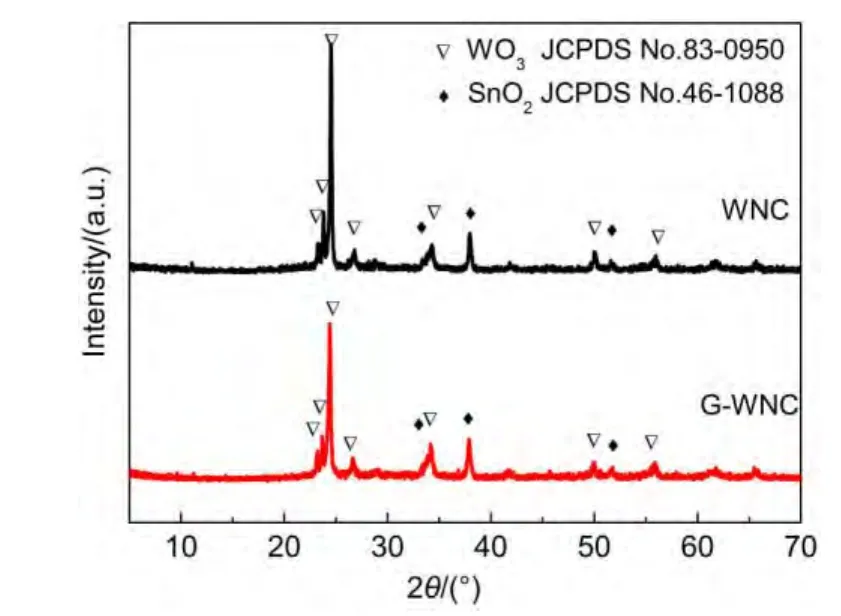
圖1 WNC和G-WNC薄膜的XRD圖譜Fig.1 XRD patterns of WO3nanocrystal(WNC)film and RGO-WNC(G-WNC)film
3.1 物相和形貌表征
將熱處理后的薄膜進行X射線衍射分析,如圖1所示.其中2θ為24.37°、26.59°、34.16°和49.93°的峰分別對應WO3的(200)、(120)、(202)和(400)晶面的衍射,這些衍射峰對應的是單斜WO3的標準卡片(JCPDS No.83-0950).同時,由于薄膜是負載在導電玻璃FTO上的,可以從XRD圖譜中看到對應于四方晶系SnO2的標準卡片(JCPDS No.46-1088)的一些峰,如圖中菱形標記所示.從圖中可以發現,WO3薄膜與復合物薄膜具有相似的峰,可以發現復合物的峰高稍低,說明復合RGO后限制了WO3納米顆粒的生長,對WO3的結晶度造成一定的影響.在G-WNC的XRD圖譜中并未發現GO和RGO的峰,一方面是由于GO在熱處理的過程中分解,20另一方面由于RGO分散在WO3納米顆粒之間,類似于無定形狀態,使其低于檢測限.10
圖2為薄膜樣品熱處理前、后的掃描電鏡及熱處理后薄膜組成的透射電鏡圖.可以看到,采用提拉法制備的復合結構薄膜表面較為平滑,有些許裂紋和空隙存在.熱處理后,薄膜呈多孔形貌,這主要是由聚乙烯吡咯烷酮和聚乙二醇熱分解所致.由圖2(c,d)中的側面掃描電鏡圖可看出,薄膜總體而言較為致密,厚度分別為0.85 μm左右.從薄膜上分別刮下少許粉體,乙醇分散后滴在銅微柵上進行透射電鏡分析,如圖2(e,f)所示.從中可以看出,構成WNC的WO3顆粒粒徑約為20 nm;G-WNC中的WO3分布在RGO表面或被RGO所包裹,說明RGO與WO3在微觀尺度下充分接觸.
3.2 拉曼表征

圖2 薄膜樣品的掃描電鏡圖(a-d)和透射電鏡圖(e-f)Fig.2 SEM(a-d)and TEM(e-f)images of film samples
為進一步證明復合物中RGO的存在,對相關樣品進行了拉曼光譜研究.從圖3(a)中可以看出,氧化鎢納米顆粒薄膜中存在一些很明顯的拉曼位移峰.其中,713.93和807.87 cm-1是對應于單斜相WO3的O―W―O伸縮振動峰,270.68和328.90 cm-1對應于O―W―O的變形振動峰,而133.08和182.03 cm-1是單斜相WO3的晶格檢測峰.在石墨烯-氧化鎢復合物的拉曼光譜中,以上的特征峰均出現了向低波數方向的數值為3 cm-1左右的變化,同時182.03 cm-1處的峰變得不太明顯,可能是由于石墨烯的存在影響了氧化鎢顆粒的形成和長大,這也從側面反映了石墨烯與氧化鎢是相互接觸的.復合物的拉曼光譜中在拉曼位移1354.32和1600.42 cm-1處出現D峰和G峰.將復合物的拉曼光譜與前驅體氧化石墨烯在1000到2000 cm-1拉曼位移范圍內進行對比,如圖3(b)所示.從中可以看出,復合RGO后,ID/IG從0.95增加到1.08,這說明前驅體GO經過攪拌燒結等過程,被打碎成更小的尺寸分散在氧化鎢納米顆粒中,即氧化石墨烯平面內水平sp2支配的碳微晶區域尺寸減少,表明GO已被還原為石墨烯.11
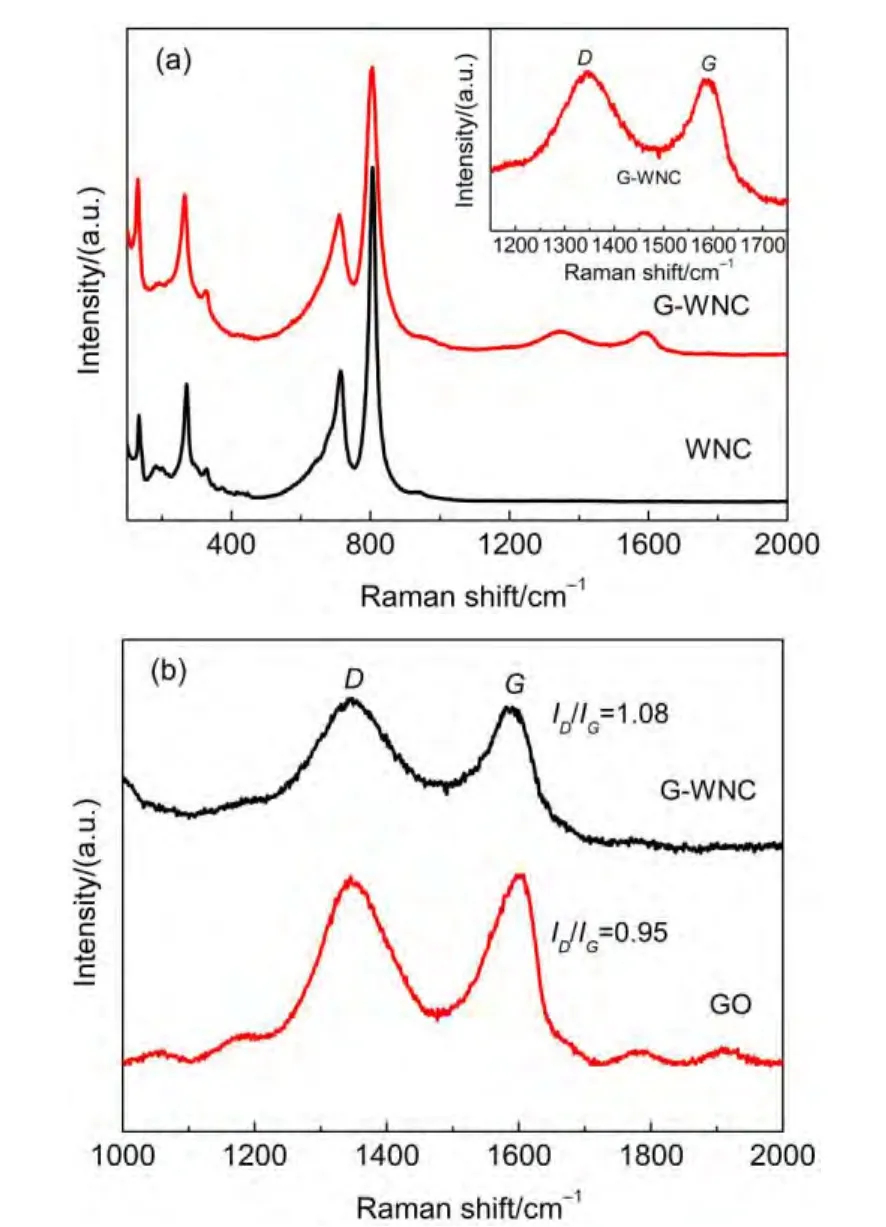
圖3 WNC和G-G-WNC(a)與GO和G-WNC(b)樣品的拉曼光譜Fig.3 Raman spectra of WNC and G-WNC(a)and graphene oxide(GO)and G-WNC(b)samples
3.3 薄膜的光電性質
圖4為不同GO含量薄膜樣品的光電流密度-電位曲線.在電位范圍為0.4-1.6 V(vsAg/AgCl)時,薄膜具有明顯的光電特性.從圖中可以看出,隨著GO添加量的增加,光電流先提升到一定值后呈現下降趨勢.這說明往氧化鎢體系中復合RGO,在一定含量范圍里對其光電轉換能力的提升效果隨RGO含量增加而增加,復合量達到一定值時體系光電轉換能力達到最佳.在本實驗中當前驅體的GO含量為28.80 mg時光電流達到最佳,為純氧化鎢薄膜的2.07倍.繼續增加RGO的含量將使薄膜電極的光電流出現下降趨勢,即光電轉換性能下降.出現這種情況的主要原因是,在低含量范圍里向半導體中添加RGO,可以利用RGO優良的導電性來提高其光生電子快速傳遞;但當RGO添加量過大時,RGO對光的吸收會降低半導體對光的有效吸收,從而影響薄膜電極整體的光電轉化能力.
為進一步研究GO對復合結構薄膜光電性能的影響,我們對薄膜進行電化學阻抗測試.如圖5所示,盡管在檢測的頻率范圍內薄膜的Nyquist圖并未形成完全的半圓形,但大多數都已出現圓弧的趨勢.從中可以看出,隨著前驅體中GO含量的增加,電極的阻抗先減小,實驗條件下GO含量為28.80 mg時阻抗最小,繼續增加前驅體中GO的添加量,阻抗則開始變大,與光電流的測試結果一致.
為了研究WNC和G-WNC薄膜的電極/溶液界面光生電荷轉移特性,本實驗在電位0.5、0.7、0.9和1.2 V(vsAg/AgCl)時對兩種電極進行了瞬態光電流實驗,如圖6所示.可以看出,在0.5 V(vsAg/AgCl)電位下,照射瞬間產生的光電流逐漸減小,并在一定時間后趨于穩定;當較高偏壓時,照射瞬間產生的光電流趨于穩定的時間變短,甚至是照射后瞬間光電流達到平衡.故瞬態光電流圖譜顯示了光電化學反應的兩個競爭過程:光生電子-空穴對的產生和復合.22光照瞬間的起始最大光電流反映了電極/溶液界面光生電子-空穴的分離,之后光電流逐漸趨于平衡的過程則說明電子-空穴的復合過程.

圖4 前驅體中不同GO含量薄膜電極的光電流曲線Fig.4 Photocurrent curves of the film electrodes with different contents of GO in the precursor
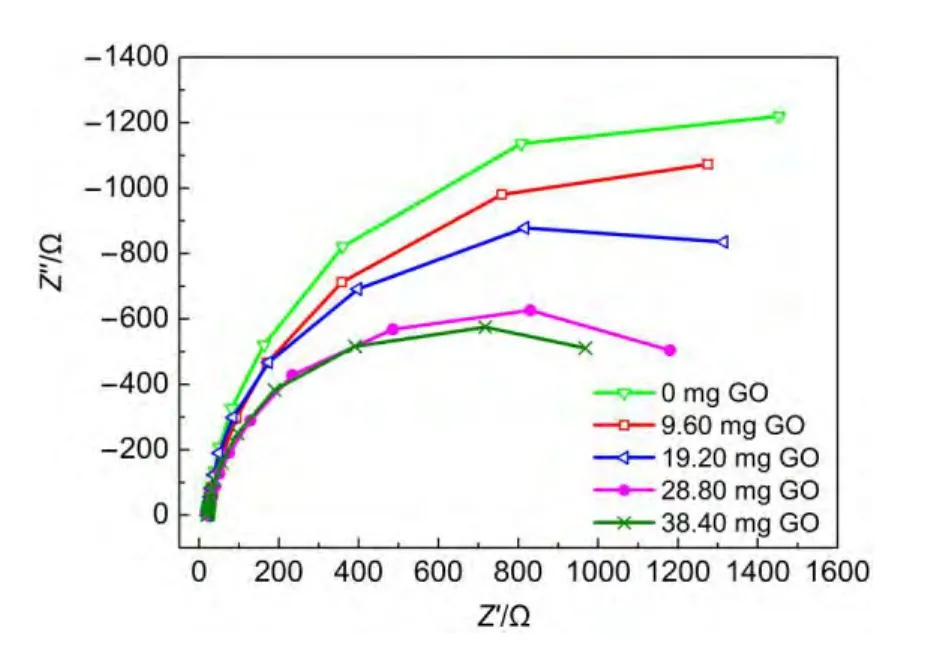
圖5 薄膜的電化學阻抗圖譜Fig.5 Electrochemical impedance spectra of the films
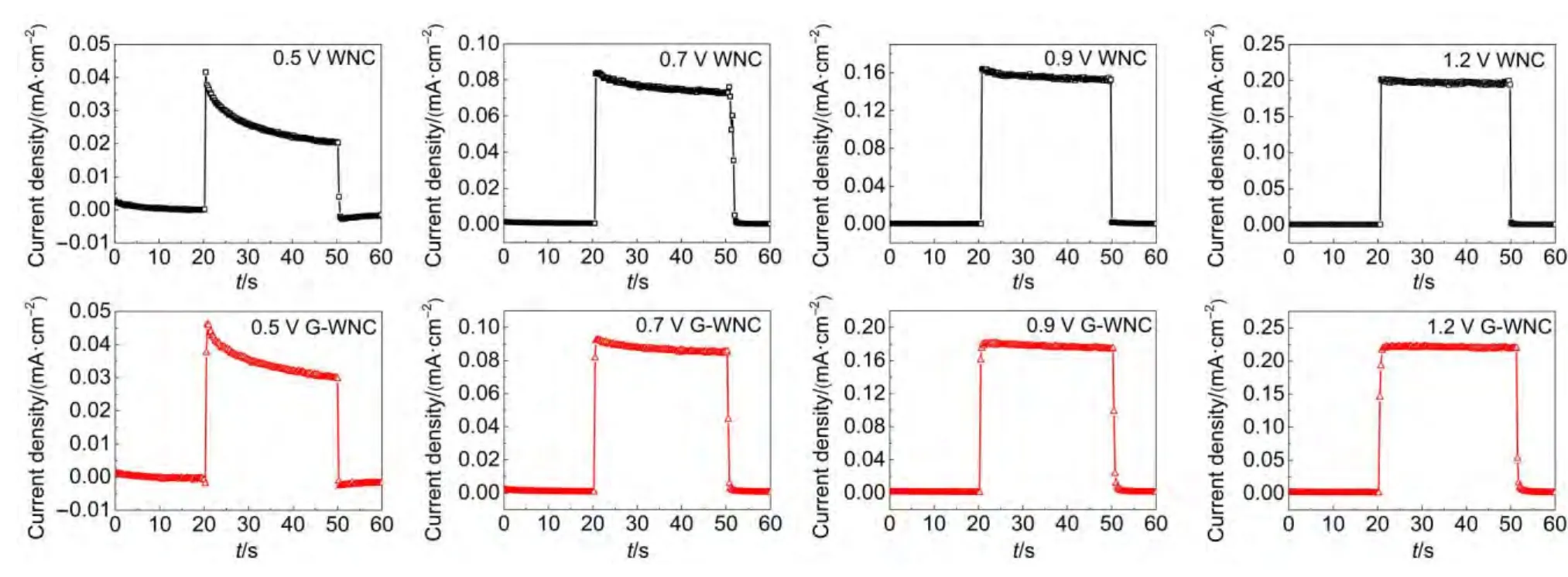
圖6 WNC與RGO G-WNC光電極在不同外加電壓下的瞬態光電流譜Fig.6 Transient photocurrent spectra of the WNC and G-WNC photoelectrodes at different applied voltages
為了進一步定量研究上述動力學行為,采用的動力學方程對光照下的瞬態光電流進行模擬,如下所示:

其中,I為光電流密度,下標i和f分別代表開始和最后,即起始光電流和光照結束時光電流;t為光照時間,It為t時刻的光電流密度;τ為瞬態時間常數,其數值可衡量光生電子-空穴對的壽命,一般定義為lnD=-1的時間t.23將不同電位下的瞬態時間常數列于表1,從中可以看出,不同電位下,相同材料的薄膜電極隨著外加電壓的增大,其瞬態時間常數增大;相同電極電位時,復合石墨烯后電極的瞬態時間常數有所增加,即電子-空穴對的壽命越長.
為了進一步研究電子的傳遞過程,采用強度調制光電流譜進行相關研究,可通過方程計算電子在薄膜中的傳輸時間τ(IMPS).24

其中fIMPS為強度調制光電流譜曲線的虛部最低點對應的頻率.在圖7中,WNC和G-WNC分別對應的是3.39和7.15 Hz,從而計算得到WNC和G-WNC薄膜中電子的傳遞時間為46.9和22.3 ms,即WNC中電子傳輸時間是G-WNC的2.1倍,這就說明石墨烯為WO3的光生電子提供有效傳輸路徑,從而提高電子傳輸速度,降低薄膜電子傳輸時間至原來的47.5%.
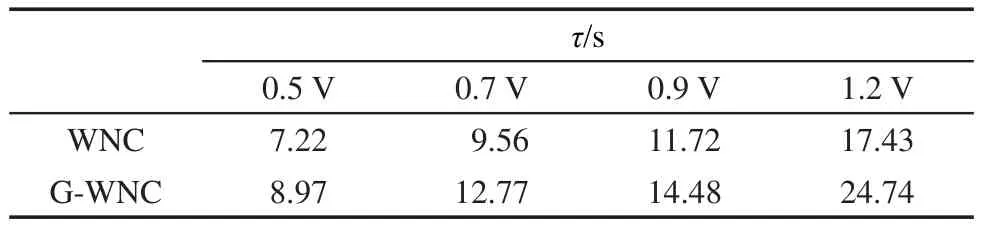
表1 WNC與G-WNC光電極在不同外加電壓下的瞬態光電流的時間常數(τ)(單位為s)Table 1 Transient time constant(τ)(unit in s)of the WNC and G-WNC photoelectrodes at different applied voltage
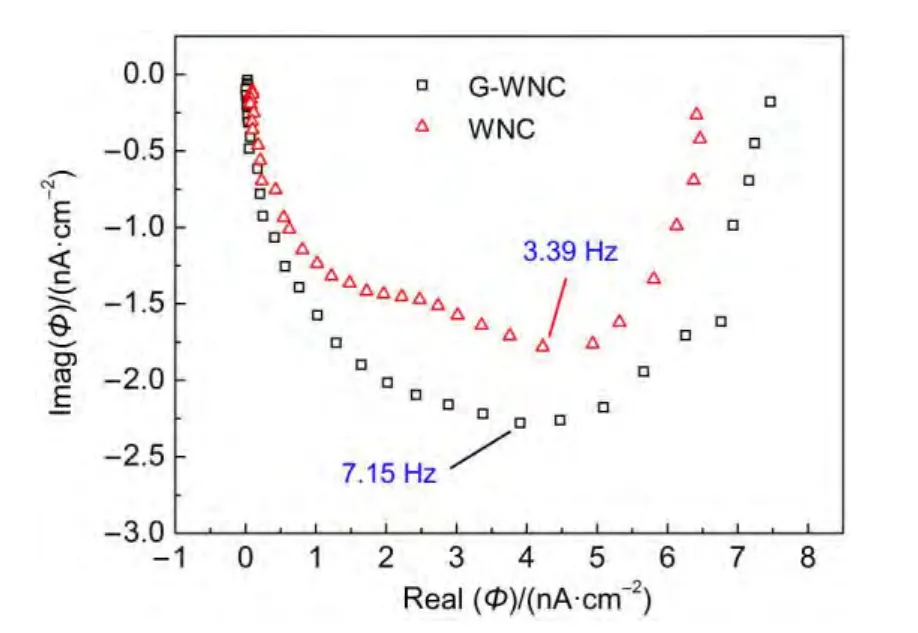
圖7 WNC與G-WNC光電極的強度調制光電流譜Fig.7 Intensity modulated photocurrent spectroscopy(IMPS)of the WNC and G-WNC photoelectrodes
4 結論
以偏鎢酸銨為鎢源、聚乙烯吡咯烷酮為連接劑,采用不同含量GO的前驅體溶液制備石墨烯-氧化鎢復合薄膜.在一定含量范圍內,前驅體中較高的GO含量有利于提高薄膜的光電轉化能力.瞬態光電流法研究表明,隨著電位的增加,WNC和GWNC的瞬態時間常數均隨之增加;在相同電極電位下,G-WNC的瞬態時間常數較WNC增大,說明石墨烯復合后電子-空穴對壽命延長.通過強度調制光電流譜發現,復合石墨烯后G-WNC薄膜的電子傳輸時間減少,為復合石墨烯前WNC薄膜的47.5%.
(1) Pan,J.H.;Lee,W.I.Chem.Mater.2006,18,847.doi:10.1021/cm0522782
(2)Sivula,K.;Formal,F.L.;Grátzel,M.Chem.Mater.2009,21,2862.doi:10.1021/cm900565a
(3) Su,J.;Guo,L.;Bao,N.;Grimes,C.A.Nano Lett.2011,11,1928.doi:10.1021/nl2000743
(4) Jayaraman,S.;Jaramillo,T.F.;Baeck,S.H.;McFarland,E.W.J.Phys.Chem.B2005,109,22958.doi:10.1021/jp053053h
(5) Cui,X.;Guo,L.;Cui,F.;He,Q.;Shi,J.J.Phys.Chem.C2009,113,4134.
(6)Xiang,Q.;Meng,G.F.;Zhao,H.B.;Zhang,Y.;Li,H.;Ma,W.J.;Xu,J.Q.J.Phys.Chem.C2010,114,2049.doi:10.1021/jp909742d
(7) Zheng,H.;Tachibana,Y.;Kalantar zadeh,K.Langmuir2010,26,19148.doi:10.1021/la103692y
(8) Chang,M.T.;Chou,L.J.;Chueh,Y.L.;Lee,Y.C.;Hsieh,C.H.;Chen,C.D.;Lan,Y.W.;Chen,L.J.Small2007,3,658.
(9) Cole,B.;Marsen,B.;Miller,E.;Yan,Y.;To,B.;Jones,K.;Al-Jassim,M.J.Phys.Chem.C2008,112,5213.doi:10.1021/jp077624c
(10)Long,M.;Cong,Y.;Li,X.K.;Cui,Z.W.;Dong,Z.J.;Yun,G.M.Acta Phys.-Chim.Sin2013,29,1344.[龍 梅,叢 野,李軒科,崔正威,董志軍,袁觀明.物理化學學報,2013,29,1344.]doi:10.3866/PKU.WHXB201303263
(11) Gan,Y.P.;Qin,H.P.;Huang,H.;Tao,X.Y.;Fang,J.W.;Zhang,W.K.Acta Phys.-Chim.Sin.2013,29,403.[甘永平,秦懷鵬,黃 輝,陶新永,方俊武,張文魁.物理化學學報,2013,29,403.]doi:10.3866/PKU.WHXB201211022
(12) Zhang,J.;Xiong,Z.;Zhao,X.S.J.Mater.Chem.2011,21,3634.doi:10.1039/c0jm03827j
(13) Li,B.;Cao,H.J.Mater.Chem.2011,21,3346.doi:10.1039/c0jm03253k
(14) Hou,Y.;Zuo,F.;Dagg,A.;Feng,P.Nano Lett.2012,12,6464.doi:10.1021/nl303961c
(15) Guo,J.;Li,Y.;Zhu,S.;Chen,Z.;Liu,Q.;Zhang,D.;Moon,W.J.;Song,D.M.RSC Advances2012,2,1356.doi:10.1039/c1ra00621e
(16)An,X.;Yu,J.C.;Wang,Y.;Hu,Y.;Yu,X.;Zhang,G.J.Mater.Chem.2012,22,8525.doi:10.1039/c2jm16709c
(17)Yang,P.;Huang,H.;Yue,Z.;Li,G.;Wang,X.;Huang,J.;Du,Y.J.Mater.Chem.A2013,1,15110.doi:10.1039/c3ta13433d
(18)Wu,H.;Xu,M.;Da,P.;Li,W.;Jia,D.;Zheng,G.Phys.Chem.Chem.Phys.2013,15,16138.doi:10.1039/c3cp53051e
(19) Tang,L.;Wang,Y.;Li,Y.;Feng,H.;Lu,J.;Li,J.Adv.Funct.Mater.2009,19,2782.doi:10.1002/adfm.v19:17
(20) Xiang,Q.;Yu,J.;Jaroniec,M.Nanoscale2011,3,3670.doi:10.1039/c1nr10610d
(21) Dang,H.;Dong,X.;Dong,Y.;Huang,J.Int.J.Hydrog.Energy2013,38,9178.doi:10.1016/j.ijhydene.2013.05.061
(22)Radecka,M.;Sobas,P.;Wierzbicka,M.;Rekas,M.Physica B:Condensed Matter2005,364,85.doi:10.1016/j.physb.2005.03.039
(23) Hagfeldt,A.;Lindstr?m,H.;S?dergren,S.;Lindquist,S.E.J.Electroanal.Chem.1995,381,39.doi:10.1016/0022-0728(94)03622-A
(24)Su,J.;Feng,X.;Sloppy,J.D.;Guo,L.;Grimes,C.A.Nano Lett.2010,11,203.
