安宮止血顆粒的質量標準研究
2014-04-11 06:28:06謝強勝
中成藥 2014年2期
王 坤, 謝強勝
(山東省食品藥品檢驗所, 山東 濟南 250101)
安宮止血顆粒的質量標準研究
王 坤, 謝強勝
(山東省食品藥品檢驗所, 山東 濟南 250101)
目的 完善安宮止血顆粒 (益母草、 馬齒莧) 的質量標準, 以控制藥品質量。 方法 修訂原標準中益母草薄層色譜法鑒別, 增加馬齒莧的薄層色譜鑒別; 采用 HPLC-ELSD法測定鹽酸水蘇堿, Venusil HILIC色譜柱, 流動相為乙腈-0.2%冰醋酸 (80 ∶20) , 體積流量 1.0 mL/min, 漂移管溫度為 90 ℃。 結果 定性鑒別方法專屬性強、 特征斑點清晰。 鹽酸水蘇堿進樣量在 1.275 ~10.20 μg范圍內, 積分值的自然對數與進樣量的自然對數線性關系良好, r= 0.999 5, 平均回收率 99.52%, RSD為 1.26%。 結論 本方法操作簡單, 結果準確, 能夠有效地控制安宮止血顆粒的質量。
安宮止血顆粒; HPLC-ELSD; 鹽酸水蘇堿
安宮止血顆粒為山東東阿阿膠股份有限公司獨家生產品種。由益母草、馬齒莧二味中藥材經過提取,濃縮至稠膏,干燥,粉碎,過篩,制成中藥顆粒劑。為了更好地控制藥品質量,提高質量標準,在原標準的基礎上修訂了薄層色譜法對益母草的鑒別, 增 加 了 馬 齒 莧 的 TLC定 性 鑒 別, 并 采 用HPLC-ELSD法, 測定益母草中鹽酸水蘇堿的量,方法簡單,靈敏,重復性好。
1 儀器與試藥
Agilent1200 高 效 液 相 色 譜 儀, Mettler AE240電子天平。 乙腈、 甲醇為色譜純 (天津康科德),無水乙醇、乙酸乙酯為分析純 (國藥集團化學試劑有限公司), 薄層色譜硅膠 G預制板 (青島海洋化工廠分廠),鹽酸水蘇堿 (購自中國藥品生物制品檢定所, 供含量測定用, 批號 110712-200508),馬齒莧對照藥材 (企業提供)。 安宮止血顆粒由山東東阿股份有限公司生產, 規格為 4g/袋; 安宮止血顆粒缺益母草陰性對照、缺馬齒莧陰性對照均由山東東阿股份有限公司提供。
2 方法與結果
2.1 益母草薄層鑒別 原標準中益母草薄層鑒別項下供試品溶液制備繁瑣[1], 展開時間較長, 本實驗對其進行驗證并進行修改。取本品 1 g或 1.5 g(加糖型), 研細,加 70%乙醇 25 mL, 超聲 30 min, 取出, 放冷, 濾過, 濾液蒸干, 殘渣加甲醇1 m L使溶解,作為供試品溶液。 另取鹽酸水蘇堿對照品, 加乙醇制成每1 mL含5 mg的溶液, 作為對照品溶液。 照薄層色譜法[1]試驗, 吸取上述兩種溶液各 10 μL, 分別點于同一硅膠 G薄層板上,以丙酮-無水乙醇-鹽酸 (10 ∶6 ∶1) 為展開劑, 展開, 取出,晾干, 在 105 ℃加熱 15 min,放冷, 噴以稀碘化鉍鉀試液-三氯化鐵試液 (10 ∶1) 混合溶液至斑點顯色清晰。供試品色譜中,在與對照品色譜相應的位置 上, 顯相同 顏色的 熒光斑 點[2-3], 陰性對照無干擾。 見圖1。

圖1 益母草的薄層色譜鑒別圖Fig.1 TLC chromatogram of Leonuri Herba
2.2 馬 齒 莧 的 薄 層 鑒 別[4-7]取 本 品 4 g或 5 g (加糖型), 研細,加稀鹽酸 3 mL與乙醇 25 mL,超聲處理 30 min, 濾過, 濾液蒸干, 殘渣加甲醇 2 mL使溶解,作為供試品溶液。另取馬齒莧對照藥材 2 g, 同法制成對 照 藥材溶液[8]。 照薄層色譜法[1]試驗, 吸取上述兩種溶液各 2 μL, 分別點于同一硅膠 G薄層板上, 以乙酸乙酯-丁酮-甲酸-水(5 ∶3 ∶1 ∶1) 為展開劑,展開,取出,晾干,噴以 0.2%茚三酮乙醇溶液,在 105 ℃加熱至斑點顯色清晰。供試品色譜中,在與對照品藥材相應的位置上,顯相同顏色的熒光斑點,陰性對照無干擾。見圖2。
2.3 鹽酸水蘇堿的測定[9-14]
2.3.1 色譜條件與系統適用性試驗 以丙基酰胺鍵合硅膠為填充劑; 乙腈-0.2%冰醋酸溶液 (80 ∶20) 為流動相; 蒸發光散射檢測器檢測; 柱溫35 ℃,體積流量 1.0 mL/min。 理論板數按鹽酸水蘇堿峰計算為 15 192。 色譜圖見圖 3。
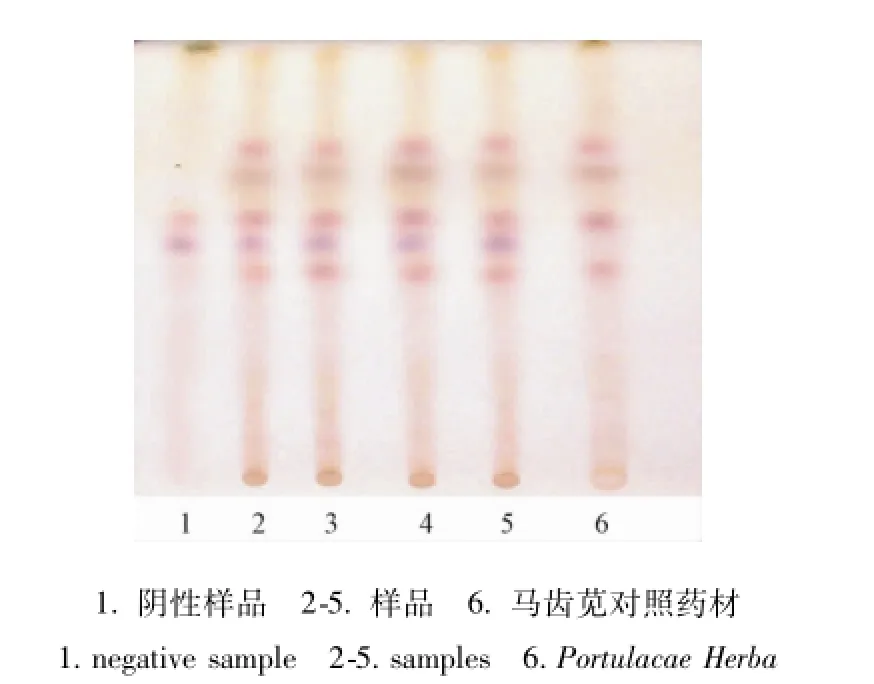
圖2 馬齒莧的薄層色譜鑒別圖Fig.2 TLC chrom atogram of Portulacae Herba
2.3.2 對照品溶液的制備 精密稱取鹽酸水蘇堿12.75 mg, 至 25 mL量瓶,加 70%乙醇定容, 搖勻, 即得 0.51 mg/mL的對照品溶液。
2.3.3 供試品溶液的制備 取裝量差異項下本品,研細, 取約 0.5 g(加糖型 0.75 g), 精密稱定, 置具塞錐形瓶中, 精密加入70%乙醇25 mL, 稱定質量, 超 聲 處 理 ( 功 率 300 W, 頻 率 40 kHz) 45 min, 取出, 放冷, 再稱定質量, 用 70%乙醇補足減失的質量,搖勻,濾過,取續濾液,即得。
2.3.4 陰性溶液的制備 取不加益母草的空白樣品按照 “2.3.3” 項下方法, 制成溶液, 即得。
2.3.5 線性關系的考察 分別精密吸取鹽酸水蘇堿對照品溶液 2.5、 5、 10、 15、20 μL, 注入液相色譜儀,測其峰面積,以峰面積的自然對數為縱坐標,對照品進樣量的自然對數為橫坐標,計算,結果回歸方程為 y=1.213 6x+7.217 3, r=0.999 5。鹽酸水蘇堿進樣量在 1.275 ~10.200 μg范圍內,進樣量自然對數與峰面積積分值自然對數呈良好的線性關系。

圖3 鹽酸水蘇堿 HPLC色譜圖Fig.3 HPLC chromatograms of stachydrine hyd rochloride
2.3.6 精密度試驗 精密吸取上述對照品溶液 10μL, 連續進樣 6 次, 測得峰面積積分值。 峰面積積分值自然對數的 RSD為 0.029%, 說明儀器精密度良好。
2.3.7 穩定性試驗 取同一批樣品按 “2.3.3”項方法制備, 分別于配制后 0、 2、 4、 6、 12 h, 精密吸取10 μL, 注入液相色譜儀, 測定峰面積, 計算峰面積積分值的自然對數及其平均值。結果鹽酸水蘇堿 RSD為 0.039%。 表明樣品在 12 h 內基本穩定。
2.3.8 重 復 性 試 驗 取 同 一 批 樣 品 ( 批 號110401)6 份,按 “2.3.3”項方法制備 6 份。 精密吸取10 μL,注入液相色譜儀, 計算,結果鹽酸水蘇堿平均含有量為 72.6 mg/袋, RSD為 1.02%。
2.3.9 加樣回收率試驗 取同一批樣品 (批號110401)6 份各 0.25 g, 精密稱定,置于具塞錐形瓶中, 分別加入對照品溶液 (鹽酸水蘇堿 1.06 mg/mL)5 m L, 再精密加入70%乙醇 20 mL, 稱定質量, 超聲處理 (功率 300 W, 頻率 40 kHz)45 min, 放冷,再稱定質量,用 70%乙醇溶液補足減失的質量,搖勻,濾過,取續濾液,即得。精密吸取 10 μL, 注入液相色譜儀, 進行測定, 計算回收率。結果見表1。
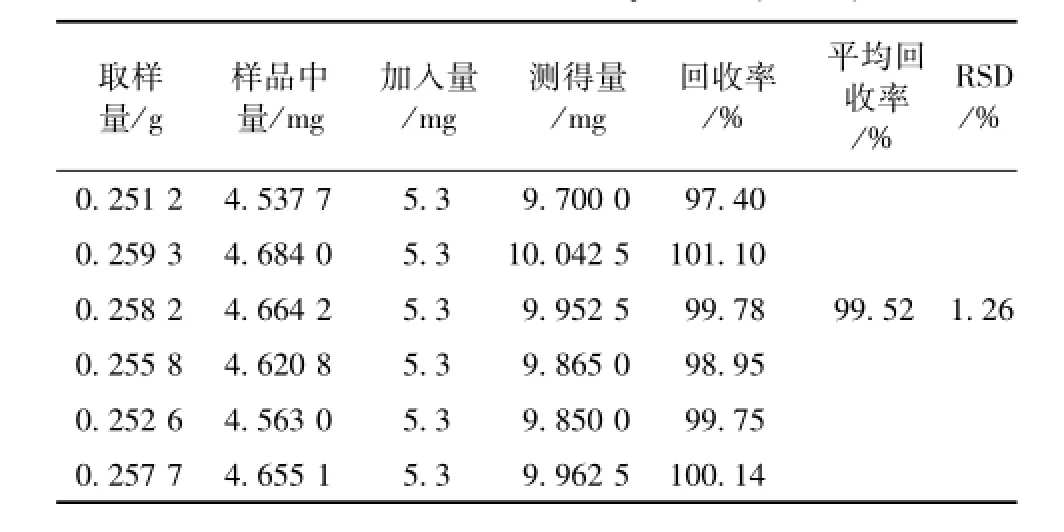
表 1 回收率試驗結果 (n=6)Tab.1 Results of recovery tests(n=6)
2.3.10 樣品測定 分別精密吸取對照品溶液 5 μL、 10 μL與供試品溶液 10 μL, 注入液相色譜儀,測定,以外標兩點法對數方程計算,即得。結果見表2。
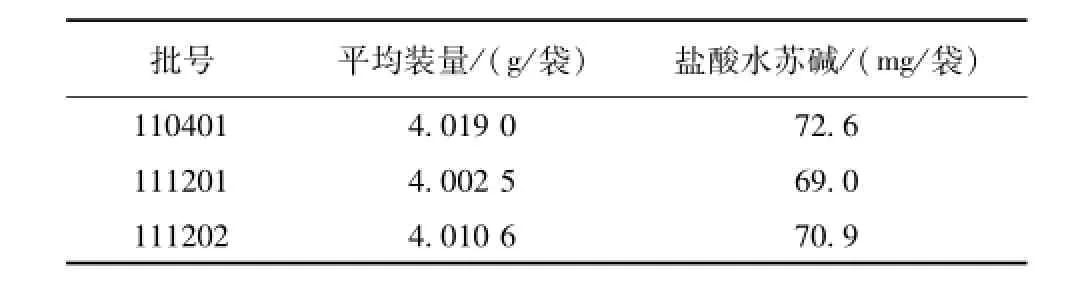
表2 鹽酸水蘇堿測定結果Tab.2 Results of the determ ination for astragaloside
3 討論
3.1 提取溶劑、 提 取方式 及取樣量考察[15-16]本實驗用甲醇、 70%乙醇、 50%乙醇作為溶媒, 超聲處理 45 min, 對安宮止血顆粒中鹽酸水蘇堿的提取率進行考察, 結果 70%乙醇提取得率高于甲醇和50%乙醇, 而且 50%乙醇提取時雜質峰較多且提取樣品不易濾過, 因此確定提取溶劑 70%乙醇。
3.2 TLC鑒別條件考察 對益母草、 馬齒莧薄層鑒別進行耐用性試驗,對不同溫濕度與不同薄層板按上述方法進行展開,均取得良好展開效果且陰性樣品無干擾,因而益母草、馬齒莧薄層色譜鑒別按上述檢測方法適用范圍廣。
[ 1 ] 國家藥典委員會.中華人民共和國藥典:2010 年版一部[S].北京: 中國醫藥科技出版社, 2010:272, 附錄ⅥB.
[2] 鄭 倩,畢雪艷, 李顯峰,等.坤寧顆粒薄層色譜鑒別[J].中國藥業, 2011, 20(3):24.
[3] 袁 敏,張貞麗.淋必通顆粒中苦地丁、益母草、綿萆薜鑒別試驗研究[J].齊魯藥事, 2012, 31(8):456-457.
[4] 徐 洋,嚴銘銘,付穎超,等.馬齒莧中總皂苷提取工藝的優選[J].時珍國醫國藥, 2010, 21(12):3104-3105.
[5] 辛海量,侯銀環,盛佳鈺,等.高效液相色譜法測定馬齒莧中不飽和脂肪酸的含量[J].解放軍藥學學報, 2011,27(1):52-54.
[6] 周 毅,曾少嫻,王成蹊,等.大孔吸附樹脂分離純化馬齒莧總黃 酮的工藝研 究[ J].時 珍國醫 國藥, 2010, 22 (3):755-757.
[7] 馮曉東,周海華,常海飛,等.延安地區馬齒莧中總黃酮的提取與含量測定 [ J].延安大學學報, 2011, 30(4): 87-89.
[ 8 ] 喬竹穩, 姚旭穎, 單喜臣, 等.馬齒莧化學成分研究[J].齊齊哈爾大學學報, 2012, 28(1):58-60.
[ 9 ] 仇其原, 鄧曉文, 吳 旸.HPLC法測定益母草膏中鹽酸水蘇堿的含量[ J] .齊魯藥事, 2010, 29(4):209-211.
[10] 朱友林.HPLC法測定癃閉舒膠囊中鹽酸水蘇堿的含量[J].中國藥房, 2011, 22(16):1509-1511.
[11] 蔣 歆, 丁永輝, 魏學冰, 等.HPLC-ELSD測定八珍益母丸中水蘇堿的含量[ J].中成藥, 2011, 33(3):542-544.
[12] 周立偉, 張 贊.HPLC測定益母草片中鹽酸水蘇堿含量[J].中國現代中藥, 2011, 13(7):33-34.
[13] 馮 旭, 杜成智, 梁臣艷, 等.HPLC-ELSD測定益母顆粒中鹽酸水蘇堿的含量 [J].中 成藥, 2008, 30(5): 163-164.
[14] 林冬杰, 張會球.HPLC-ELSD法測定益母草流浸膏中鹽酸水蘇堿的含量[J].亞太傳統醫藥, 2009, 5(2):45-46.
[15] 楊曉云, 雪 濤.益母丸中鹽酸水蘇堿成分分析方法的建立[ J].中國現代應用藥學雜志, 2008, 25(1):69-70.
[16] 陳劍鋒, 陳學松.益母草顆粒的質量標準[J].中國藥師,2009, 12(11):1551-1552.
Quality standard for Angong Zhixue G ranules
WANG Kun, XIE Qiang-sheng
(Shandong Provincial Institute for Food and Drug Control, Jinan 250101, China)
Angong Zhixue Granules; HPLC-ELSD;stachydrine hydrochloride
927.2
:A
:1001-1528(2014)02-0318-03
10.3969/j.issn.1001-1528.2014.02.022
2013-03-07
王 坤 (1971—), 男, 副主任藥師, 主要從事藥物檢驗研究。 Tel:(0531)81216510
[17] 焦 燕, 王英鋒, 劉鎖蘭.LC-MS/MS 法同時測定不同產地半枝蓮中野黃芩苷和芹菜素含量[J].藥物分析雜志,2009, 29(9):1451-1453.
