對甲苯磺酸摻雜聚苯胺材料的導電性能及其機理研究
2014-04-13 04:06:20駱巧玲阮明珠
中國塑料 2014年4期
關鍵詞:影響
吳 唯,駱巧玲,阮明珠
(華東理工大學材料科學與工程學院,中德先進材料聯合研究中心,上海200237)
0 前言
導電聚合物因其優良的導電和光學性能而在電池[1]、電致變色器件[2]、傳感器[3]、防腐器件[4]、發光二極管[5]、抗靜電[6]等領域廣泛應用。PANI被認為最有希望獲得實際應用的導電聚合物,因其具有容易制備[7]、良好的環境穩定性、較高的電導率和優異的光學性能[8]。現已被大家公認的PANI的結構是MacDiarmid等[9]提出的結構式如圖1所示,式中y 值在0和1之間。不同的y 值可對應于3 種不同的PANI穩定態:全還原態y=1,中間氧化態y=0.5 和全氧化態y=0,各態之間可以相互轉變。目前關于PANI的研究都集中于其中間氧化態,也稱本征態,這不僅因為該態穩定,更主要的是通過質子酸摻雜可使其具有導電性。質子酸摻雜就是H+與共軛聚合物作用而使導電性增加的過程,經摻雜后共軛聚合物的導電性往往會增加幾個數量級,有機功能磺酸摻雜PANI可以同時大幅度地提高電導率和熱穩定性,故有機磺酸摻雜PANI成為研究的熱點。
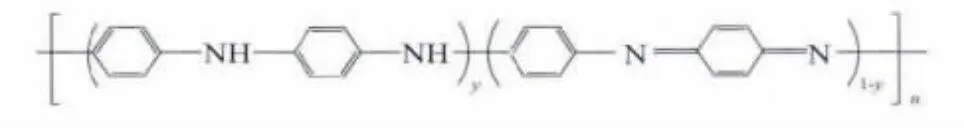
圖1 PANI的結構式Fig.1 Structural formula of PANI
本文采用TSA 經再摻雜制得摻雜態PANI,測其電導率,分析摻雜時間和TSA 水溶液濃度對其電導率的影響;采用FTIR 分析摻雜前后PANI的結構變化,探究摻雜時間對摻雜態PANI導電性影響的機理;用TG 分析TSA 對PANI熱穩定性的影響。
1 實驗部分
1.1 主要原料
本征態PANI,純度99%,石家莊冀安亞大新材料科技有限公司;
TSA,分析純,上海凌峰化學試劑有限公司。
1.2 主要設備及儀器
數字式四探針測試儀,SZ-82,蘇州電訊儀器廠;
TG,WRT-2P,上海精科天平儀器廠。
1.3 樣品制備
在裝有本征態PANI的圓底燒瓶中,加入一定濃度的TSA 水溶液,使苯胺與TSA 的摩爾比為1∶0.8,在25 ℃恒溫水浴,恒定轉速條件下反應一段時間,反應所得產物經過濾洗滌至濾液無色后,在60 ℃及真空條件下干燥24h,得到摻雜態PANI。
1.4 性能測試與結構表征
FTIR 分析:采用KBr壓片,測試波數范圍4000~500cm-1;
電導率測定:取適量摻雜態PANI,在室溫、10 MPa壓力下壓制成圓片試樣進行測試;
TG 分析:空氣氣氛,以10 ℃/min 升溫速率從30 ℃升到700 ℃,記 錄TG 曲線。
2 結果與討論
2.1 摻雜時間對PANI性能的影響
2.1.1 摻雜時間對PANI導電性能影響
圖2為摻雜時間對摻雜態PANI的電導率影響,其中TSA 水溶液的濃度為0.5 mol/L,本征態PANI的電導率非常低,只有1.03×10-6S/cm,但經TSA 摻雜,PANI的電導率從10-6S/cm 增加到10-2S/cm,增大了4個數量級,這與無機酸摻雜PANI的電導率相近[10],說明TSA 可明顯提高PANI的電導率。隨摻雜時間的延長,摻雜態PANI的電導率先增大后減小,在摻雜時間為12h時電導率達最大值0.096S/cm。
2.1.2 不同摻雜時間PANI的FTIR分析
大興安嶺不同林草過渡區土壤碳庫活度的研究……… 田 慧,包 翔,周 梅,趙鵬武,石 亮,巴音德樂黑,郝良杰,烏藝恒(47)
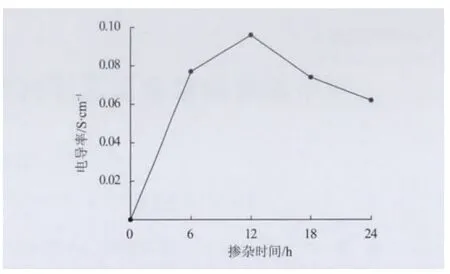
圖2 摻雜時間對PANI/TSA 的電導率的影響Fig.2 Effect of doping time on conductivity of PANI/TSA
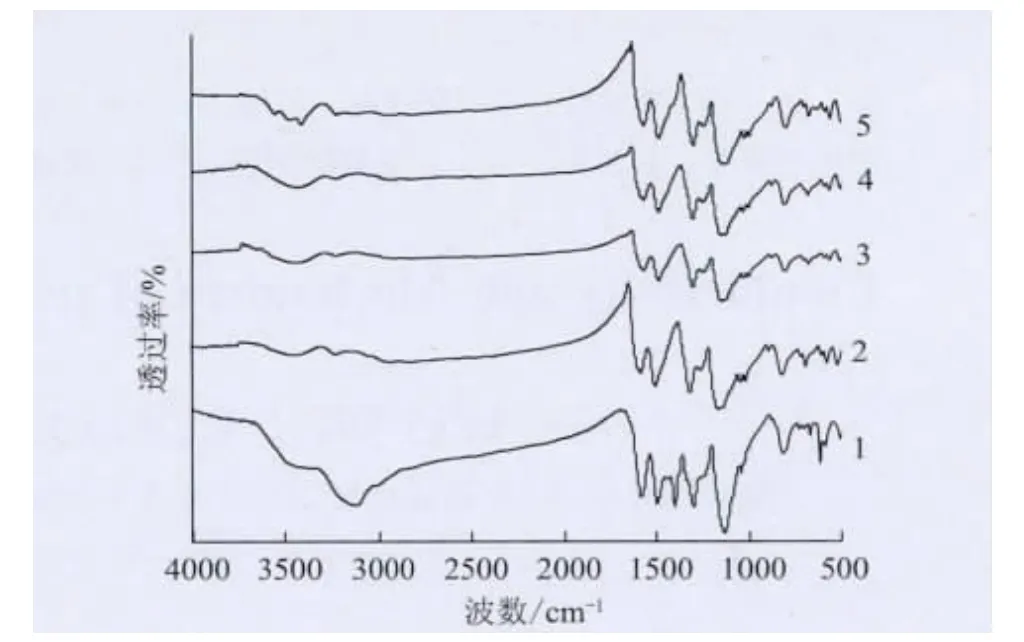
圖3 不同摻雜時間的PANI/TSA 的FTIR 譜圖Fig.3 FTIR of PANI/TSA with different doping time
圖3為本征態PANI和不同摻雜時間的TSA 摻雜PANI的FTIR 譜圖,其中TSA 水溶液的濃度為0.5mol/L。可以看出,與本征態PANI(曲線1)相比,經TSA 摻雜的PANI(曲線2~5)均出現了 S═ O 鍵伸縮振動吸收峰,且當摻雜時間為6、12、18、24h 時,PANI/TSA 的 S ═O 鍵伸縮振動吸收峰分別出現在1029cm-1和1005cm-1、1029cm-1和1005cm-1、1032cm-1和1006cm-1、1028cm-1和1004cm-1處。S ═O 鍵伸縮振動吸收峰是TSA 上磺酸基團的特征峰[11],上述結果說明TSA成功摻雜到PANI分子鏈上。
圖3還顯示,曲線1在1400cm-1處存在一個苯醌單元(Q ═ N—B)的伸縮振動峰,但曲線2~5上該峰消失,說明PANI經TSA摻雜后,TSA中的H+與 Q═ N—B中的N結合后變成了Q—N—B基團。正是這一變化,導致電子由基本定域在氮原子上變成在一定的共軛長度范圍內分布,從而提高了PANI的電導率。
另外由圖3還發現,經TSA 摻雜的PANI的特征吸收峰多數向低波數移動了大約10cm-1,這很可能是由于亞胺氮質子化使聚合物分子鏈的電子云密度下降,降低了原子間的力常數,使電子躍遷所需要的能量降低,導致功能磺酸摻雜PANI的FTIR 譜圖發生紅移[12]。但正是亞胺氮質子化,促進了電子由基本定域在氮原子上變成在一定的共軛長度范圍內分布,提高分子鏈的共軛程度,從而提高了PANI的電導率。
2.1.3 摻雜時間對PANI導電性影響的機理分析
TSA 上的H+使亞胺氮質子化,導致亞胺氮帶上正電荷,此正電荷在共軛鏈上延展離域到鄰近苯環及其對位N 原子上,使分子鏈中N 原子的化學環境產生了一定程度的均化,從而提高了電導率[13]。而摻雜與脫摻雜是一個可逆競爭過程,這一過程可如圖4 摻雜原理描述:摻雜過程剛開始時,TSA 先是包覆在PANI表面,并開始與亞胺氮形成氫鍵,此時摻雜過程占主導;隨時間的延長,PANI上的質子氫逐漸增多,摻雜與脫摻雜趨向平衡,此時摻雜態PANI的電導率最大;繼續延長時間,則再摻雜過程中形成的N—H 氫鍵經過長時間的攪拌斷裂,且反應產物可能發生水解,使電導率下降[14]。
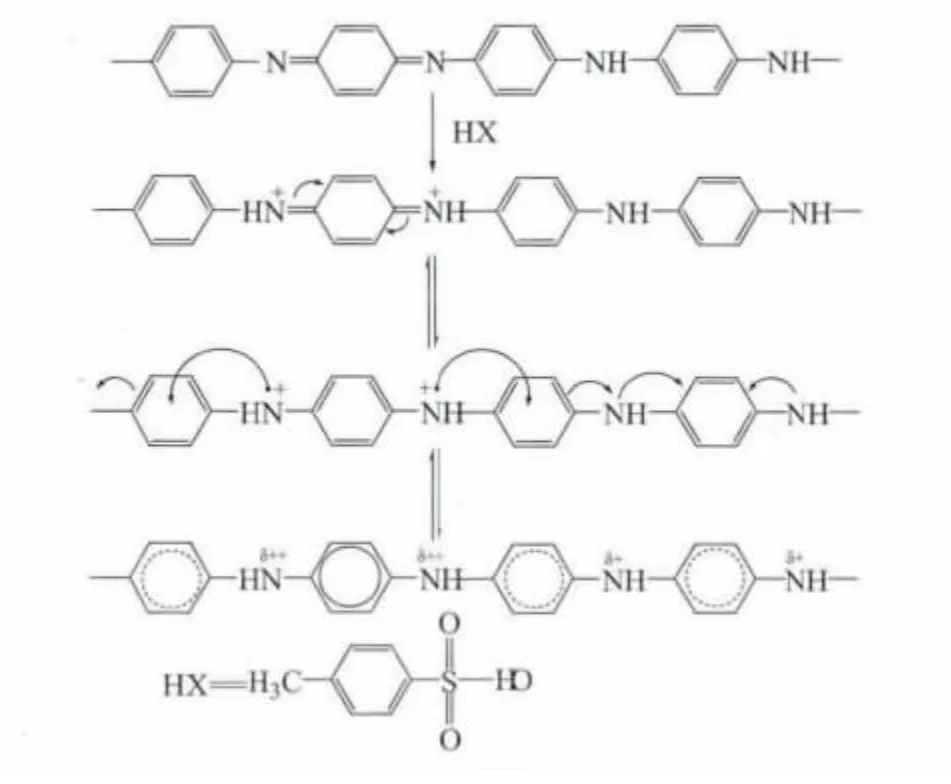
圖4 TSA 摻雜PANI原理Fig.4 Mechanism of p-toluene sulfonic acid doping polyaniline
2.2 TSA 濃度對PANI性能的影響
2.2.1 TSA 濃度對PANI導電性能影響
圖5為TSA 濃度對摻雜態PANI的電導率影響(摻雜時間均為12h)。可以發現,TSA 水溶液濃度為0.5mol/L 時,PANI/TSA 的 電導率為0.096S/cm,TSA 水溶液濃度增大到1.0mol/L 時,PANI/TSA 的電導率為0.081S/cm,均比PANI(1.03×10-6S/cm)電導率增大4個數量級,說明TSA 可明顯提高PANI的電導率。TSA 水溶液濃度為0.5 mol/L 的PANI/TSA 的電導率大于TSA 水溶液濃度為1.0mol/L 的PANI/TSA 的電導率,TSA 濃度的增大并沒有引起PANI電導率的增大[15],這是由于加入TSA 質量相同的前提下,TSA 水溶液濃度小可提高TSA 與PANI粉末接觸的概率,從而提高N—H 鍵形成的概率,故可提高PANI/TSA 的電導率,但TSA 水溶液濃度進一步減小,會導致溶液/PANI質量過大,導致生產效率降低,不利于PANI/TSA 的大量制備。
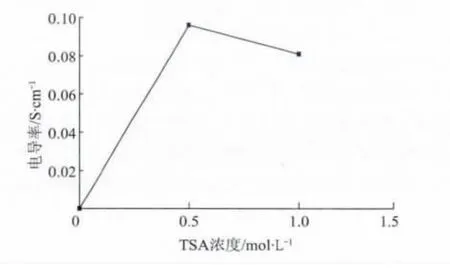
圖5 TSA 濃度對PANI/TSA 電導率的影響Fig.5 Effect of TSA concentration on conductivity of PANI/TSA
2.2.2 不同TSA濃度PANI的FTIR分析
圖6 為本征態PANI和不同TSA 濃度的摻雜PANI的FTIR 譜圖(摻雜時間均為12h)。可以看出,經TSA 摻雜后,摻雜態PANI(曲線2和3)上都出現了本征態PANI(曲線1)不存在的 S═ O 鍵伸縮振動吸收峰(1029cm-1和1005cm-1),該峰屬于TSA 的特征峰。而本征態PANI存在的Q═ N—B 中的C—N 伸縮振動峰(曲線1,1400cm-1)在摻雜后消失。這很可能是TSA 中的H+與亞胺氮形成氫鍵,導致醌環消失之故。另外,圖6還顯示,2種TSA 濃度摻雜的PANI與本征態PANI相比,其特征吸收峰(1578cm-1和1496cm-1)發生大約10cm-1紅移,這可解釋為因亞胺氮質子化,降低了N 原子的電子密度,但促進了電子由基本定域在氮原子上變成在一定的共軛長度范圍內分布,提高分子鏈的共軛程度,從而提高了PANI的電導率。但不同濃度的TSA 水溶液濃度對PANI特征吸收峰紅移的影響較小。
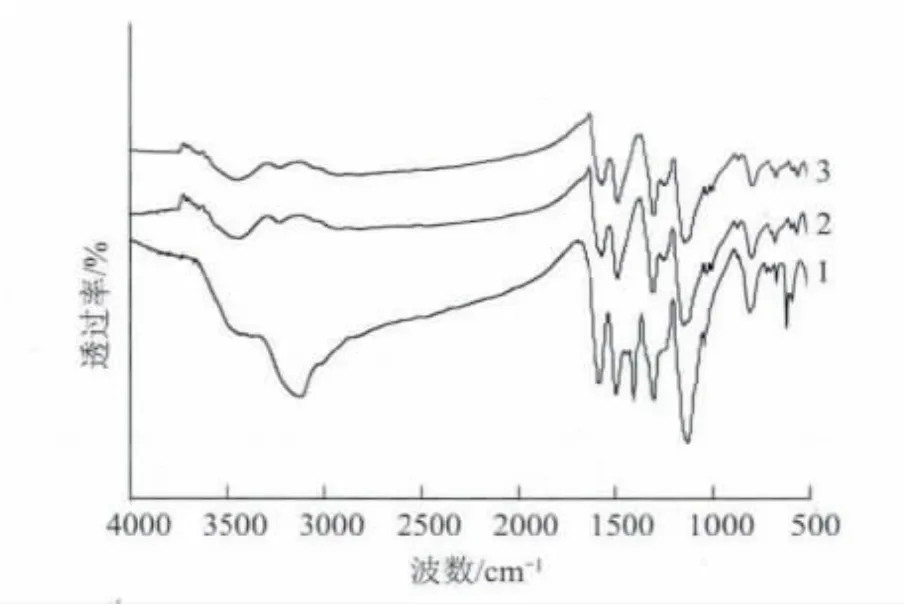
圖6 不同TSA 濃度的PANI/TSA 的FTIR 譜圖Fig.6 FTIR of PANI/TSA with different TSA concentration
2.3 摻雜時間和TSA 濃度對PANI熱穩定性的影響
圖7 為本征態PANI和不同摻雜時間的摻雜態PANI的TG 曲線,表1 是相關的TG 數據。圖7 顯示,本征態PANI的起始分解溫度為202 ℃,其代表PANI的氧化及分解。而TSA 摻雜的PANI都出現了2個明顯的失重峰,第一個失重峰代表的是TSA 根離子的分解及氧化,第二個失重峰代表的是PANI的氧化及分解。這2個失重峰溫度都明顯高于本征態PANI,說明經TSA 摻雜后PANI熱穩定性得到提高。
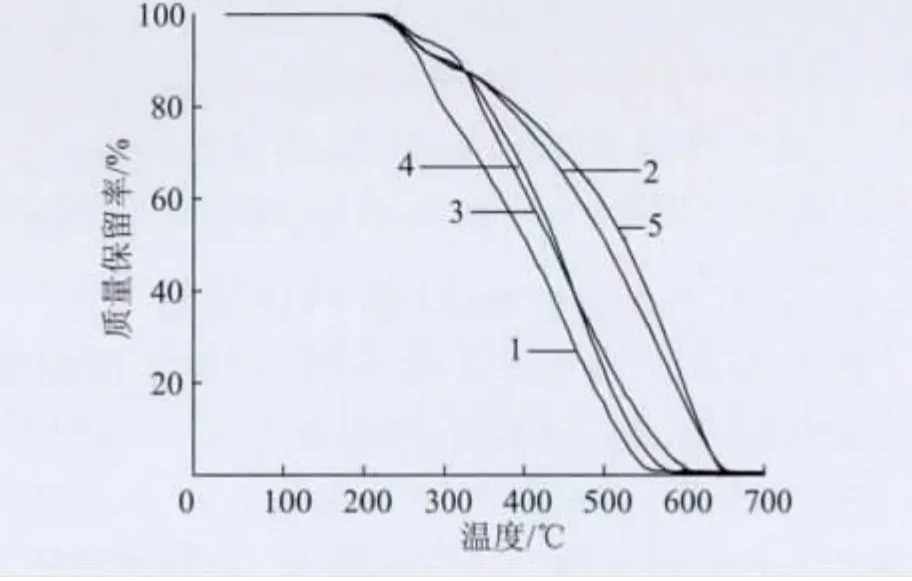
圖7 不同摻雜時間的PANI/TSA 的TG 曲線Fig.7 TG curves of PANI/TSA with different doping time
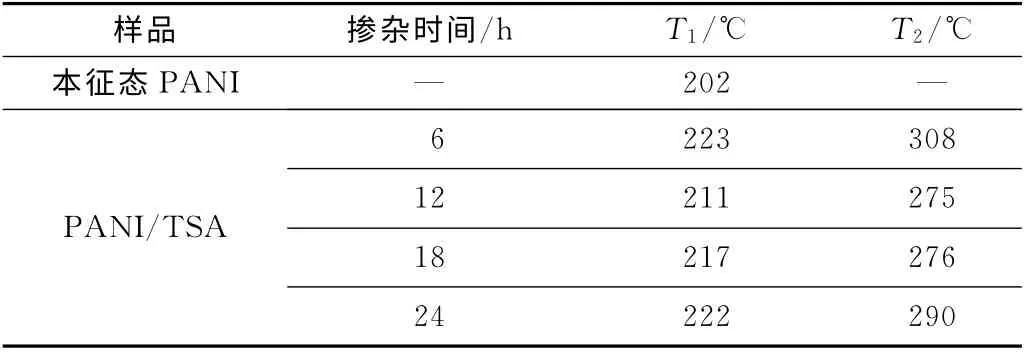
表1 不同摻雜時間的摻雜態PANI的起始分解溫度Tab.1 Initial decomposition temperature of PANI/TSA with different doping time
注:T1——第一個分解峰起始溫度;T2——第二個分解峰起始溫度。
圖8為本征態PANI和不同TSA 濃度的摻雜態PANI的TG 曲線,表2是其TG 數據。結果顯示,不同TSA 濃度的PANI/TSA 的熱穩定性同樣都比本征態PANI高。
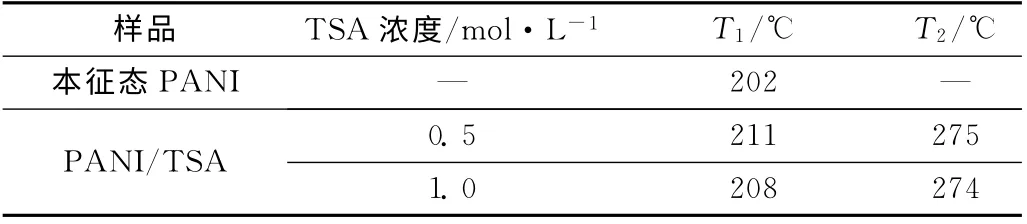
表2 不同TSA濃度的PANI/TSA的起始分解溫度Tab.2 Initial decomposition temperature of PANI/TSA with different TSA concentration
3 結論
(1)采用再摻雜法成功制備了TSA 摻雜PANI,摻雜態PANI的電導率明顯提高;
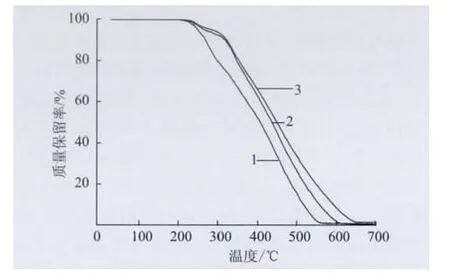
圖8 不同TSA 濃度的PANI/TSA 的TG 曲線Fig.8 TG curves of PANI/TSA with different TSA concentration
(2)TSA 與PANI的摻雜是一個摻雜與脫摻雜的可逆競爭過程,隨摻雜時間的延長,摻雜態PANI的電導率先增大后減小,在摻雜時間為12h時電導率達最大值0.096S/cm;
(3)不同TSA 水溶液濃度的TSA 均可提高PANI的電導率,但隨TSA 濃度的增大,電導率有所下降,摻雜時間為12h,TSA 濃度為0.5mol/L 制得的摻雜態PANI具有最好的電導率(0.096S/cm)。
[1] Li Duan,Jiachun Lu,Wenyuan Liu.Fabrication of Conductive Polymer-coated Sulfur Composite Cathode Materials Based on Layer-by-layer Assembly for Rechargeable Lithiumsulfur Batteries[J].Colloids and Surfaces A:Physicochemical and Engineering Aspects,2012,414:98-103.
[2] Eliana A R,De Paoli M A,Mastragostino M.An Electrochromic Device Based on Polyaniline and Prussian Blue[J].Advanced Materials,1992,4(4):287-291.
[3] Sotomayor M D T,DePaoli M A,DeOliveira W A.Fiberoptic pH Sensor Based on Poly(o-methoxy-aniline)[J].Analytica Chimica Acta,1997,353(2):275-280.
[4] Wessling B.Passivation of Metals by Coating with Polyaniline:Corrosion Potential Shift and Morphological Changes[J].Advanced Materials,1994,3(6):226-228.
[5] Yuh-Ruey Y,Hsia-Tsai H,Chun-Guey W.The Application of Insitu Prepared Polyaniline Film as a Hole Blocking Layer in Polymeric Organic Light Emitting Diode[J].Synthetic Metals,2001,121:1651-1652.
[6] John D.Stenger Smith.Intrinsically Electrically Conducting Polymers.Synthesis,Characterization,and Their Applications[J].Progress in Polymer Science,1998,23(1):57-59.
[7] Ray S,Easteal A J,Cooney R P.et al.Structure and Properties of Melt-processed PVDF/PMMA/Polyaniline Blends[J].Materials Chemistry and Physics,2009,113(2):829-838.
[8] Mitzakoff S,Paoli M-A D.Blends of Polyaniline and Engineering Plastics[J].European Polymer Journal,1999,35:1791-1798.
[9] MacDiarmid A G,Epstein A J.Polyaniline:A Novel Concept in Conducting Polymer[J].Synthetic Metals,1987,18:285.
[10] 鐘新仙,馮崎鵬,黃有國,等.超級電容器用摻雜聚苯胺納米材料的乳液聚合法及電容性能研究[J].功能材料,2013,44(19):2800-2809.Zhong Xinxian,Peng Qipeng,Huang Youguo et al.Research on Emulsion Polymerization Method and Capacitive Performance of Doped PANI Nanomaterial for Supercapacitor[J].Journal of Functional Materials,2013,44(19):2800-2809.
[11] Mu Yang,Zhaojun Xiang,Ge Wang.A Novel Orchidlike Polyaniline Superstructrue by Solvent-thermal Method[J]Journal of Colloid and Interface Science,2012,367:49-54.
[12] Yuan Tongsuo,Huang Yan,Dong Shujuan,et a1.Infrared reflection of Conducting Polyaniline Polymer Coating[J].Polymer Testing,2002,21:641-646.
[13] 吳 丹,朱 超,強驥鵬,等.聚苯胺的摻雜及其應用[J].工程塑料應用,2006,34(9):70-73.Wu Dan,Zhu Chao,Qiang Jipeng,et al.Doping and Application of Polyaniline[J].Engineering Plastics Application,2006,34(9):70-73.
[14] 王雅珍,張 巖,許嘉航.二次摻雜聚苯胺的PAN 復合纖維抗靜電性能研究[J].合成纖維工業,2013,36(1):10-12.Wang Yazhen,Zhang Yan,Xu Jiahang.Antistatic Property of PAN Composite Fiber After the Second Doping[J].China Synthetic Fiber Industry,2013,36(1):10-12.
[15] Perez-Martinez C J,Castillo-Castro T,Castillo-Ortega M M,et al.Preparation of Polyaniline Submicro/Nanostrucutres Using L-glutamic Acid:Loading and Releasing Studies of Amoxicilin[J].Synthetic Metals,2013,184:41-47.
猜你喜歡
中學生數理化·八年級物理人教版(2022年3期)2022-03-16 05:55:08
當代陜西(2021年2期)2021-03-29 07:41:24
家庭影院技術(2020年10期)2020-12-14 07:54:18
媽媽寶寶(2017年3期)2017-02-21 01:22:28
中國塑料(2016年3期)2016-06-15 20:30:00
通信電源技術(2016年3期)2016-03-26 07:13:38
知識經濟·中國直銷(2016年3期)2016-02-27 16:15:49
現代檢驗醫學雜志(2014年6期)2014-02-02 03:02:04
閱讀與作文(小學低年級版)(2011年3期)2011-01-01 00:00:00
